This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Supercomputing simulations spot electron orbital signatures
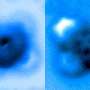
 surface. a Experimental constant-height AFM frequency-shift images (V = 0 V, tip amplitude = 100 pm) using a CO tip at a tip height of −10 pm with respect to our 100 mV/100 pA STM set point. The two white dashed circles highlight the main differences between these two molecules—the central metal atom. b Glow-edges filtered experimental AFM image (based on a). c Simulated AFM images with a CO tip at a tip height of −10 pm (see Supplementary Information for the definition of tip height in simulation). Left panel: spin-polarized DFT calculations; right panel: spin-paired DFT calculations (indicated by a superscript *). On the midline, the orbital-like figures are the calculated total electron density differences between MPc and M*Pc (MPc–M*Pc). Yellow: positive, cyan: negative. Isovalue: 0.003 e–/bohr3. d Estimated width (in pm) of the central part of the MPcs based on the signal strength—I value. The white dashed arrow pointing from b to d indicates a zoomed-in image of the central part of the left FePc molecule. The white curves are calculated I values along the corresponding dashed axes. The blue arrows illustrate how we define the width of the square based on I values. Top panel: FePc (in blue), bottom panel: CoPc (in red). Each MPc has two widths and corresponds to two circles. The gap between the two dashed black lines (the highest red and lowest blue circles) shows a minimum difference of 30 pm. Credit: Nature Communications (2023). DOI: 10.1038/s41467-023-37023-9")
No one will ever be able to see a purely mathematical construct such as a perfect sphere. But now, scientists using supercomputer simulations and atomic resolution microscopes have imaged the signatures of electron orbitals, which are defined by mathematical equations of quantum mechanics and predict where an atom's electron is most likely to be.
Scientists at UT Austin, Princeton University, and ExxonMobil have directly observed the signatures of electron orbitals in two different transition-metal atoms, iron (Fe) and cobalt (Co) present in metal-phthalocyanines. Those signatures are apparent in the forces measured by atomic force microscopes, which often reflect the underlying orbitals and can be so interpreted.
Their study was published in March 2023 as an Editors' Highlight in the journal Nature Communications.
"Our collaborators at Princeton University found that despite Fe and Co being adjacent atoms on the periodic table, which implies similarity, the corresponding force spectra and their measured images show reproducible experimental differences," said study co-author James R. Chelikowsky, the W.A. "Tex" Moncrief, Jr. Chair of Computational Materials and professor in the Departments of Physics, Chemical Engineering, and Chemistry in the College of Natural Sciences at UT Austin. Chelikowsky also serves as the director of the Center for Computational Materials at the Oden Institute for Computational Engineering and Sciences.
Without a theoretical analysis, the Princeton scientists could not determine the source of the differences they spotted using high-resolution non-contact atomic force microscopy (HR-AFM) and spectroscopy that measured molecular-scale forces on the order of piconewtons (pN), one-trillionth of a Newton.
"When we first observed the experimental images, our initial reaction was to marvel at how experiment could capture such subtle differences. These are very small forces," Chelikowsky added.
"By directly observing the signatures of electron orbitals using techniques such as atomic force microscopy, we can gain a better understanding of the behavior of individual atoms and molecules, and potentially even how to design and engineer new materials with specific properties. This is especially important in fields such as materials science, nanotechnology, and catalysis," Chelikowsky said.
The required electronic structure calculations are based on density functional theory (DFT), which starts from basic quantum mechanical equations and serves as a practical approach for predicting the behavior of materials.
"Our main contribution is that we validated through our real-space DFT calculations that the observed experimental differences primarily stem from the different electronic configurations in 3d electrons of Fe and Co near the Fermi level, the highest energy state an electron can occupy in the atom," said study co-first author Dingxin Fan, a former graduate student working with Chelikowsky. Fan is now a postdoctoral research associate at the Princeton Materials Institute.
The DFT calculations included the copper substrate for the Fe and Co atoms, adding a few hundred atoms to the mix and calling for intense computation, for which they were awarded an allocation on the Stampede2 supercomputer at the Texas Advanced Computing Center (TACC).
"In terms of our model, at a certain height, we moved the carbon monoxide tip of the AFM over the sample and computed the quantum forces at every single grid point in real space," Fan said. "This entails hundreds of different computations. The built-in software packages on TACC's Stampede2 helped us to perform data analysis much more easily. For example, the Visual Molecular Dynamics software expedites an analysis of our computational results."
"Stampede2 has provided excellent computational power and storage capacity to support various research projects we have," Chelikowsky added.
By demonstrating that the electron orbital signatures are indeed observable using AFM, the scientists assert that this new knowledge can extend the applicability of AFM into different areas.
What's more, their study, used an inert molecular probe tip to approach another molecule and accurately measured the interactions between the two molecules. This allowed the science team to study specific surface chemical reactions.
For example, suppose that a catalyst can accelerate a certain chemical reaction, but it is unknown which molecular site is responsible for the catalysis. In this case, an AFM tip prepared with the reactant molecule can be used to measure the interactions at different sites, ultimately determining the chemically active site or sites.
Moreover, since the orbital level information can be obtained, scientists can gain a much deeper understanding of what will happen when a chemical reaction occurs. As a result, other scientists could design more efficient catalysts based on this information.
Said Chelikowsky: "Supercomputers, in many ways, allow us to control how atoms interact without having to go into the lab. Such work can guide the discovery of new materials without a laborious 'trial and error' procedure."
More information: Pengcheng Chen et al, Observation of electron orbital signatures of single atoms within metal-phthalocyanines using atomic force microscopy, Nature Communications (2023). DOI: 10.1038/s41467-023-37023-9
Journal information: Nature Communications
Provided by University of Texas at Austin