This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
proofread
Researchers explain origins of dangerous coronavirus variants
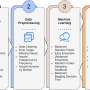
 Clusters of concordantly evolving sites. Graph vertices represent sites, and edges represent concordantly evolving pairs (Appendix 1—table 7). The graph consists of 13 connected components, 8 of which contain just a single edge. Site pairs that were among the set of the best scoring pairs predicted for the alternative UShER (Turakhia et al., 2021) topology (FDR <10%, Table 1) are marked by asterisks. (B) Concordantly evolving sites among the lineage-defining sites of Alpha, Beta, Gamma, Delta (B.1.617.2+AY.*) and Omicron (BA.1) VOCs (Hodcroft, 2021). Concordantly evolving sites are colored in accordance with the clusters in panel A. Sites with fewer than 2 mutations which were not included in the analysis are in gray. Credit: eLife (2023). DOI: 10.7554/eLife.82516")
HSE researchers, in collaboration with their colleagues from Skoltech and the Central Research Institute for Epidemiology, have uncovered the mechanisms behind the emergence of new and dangerous coronavirus variants, such as alpha, delta, omicron, and others. They have discovered that the likelihood of a substitution occurring at a specific site of the SARS-CoV-2 genome is dependent on concordant substitutions occurring at other sites.
This explains why new and more contagious variants of the virus can emerge unexpectedly and differ significantly from those that were previously circulating. The study's findings have been published in eLife.
All viruses, including the SARS-CoV-2 virus responsible for the coronavirus pandemic, change over time. The longer a virus circulates within the population and the more individuals it infects, the more changes (mutations) it accumulates. Typically, new strains of a virus are similar to their parental strains.
However, in some cases, mutations may result in variants that are better adapted to the environment and pose a higher risk to human health. These variants tend to spread more quickly, may not respond to existing vaccines or treatments, and could be more difficult to diagnose.
Throughout the history of the coronavirus, there have been various instances of highly active variants unexpectedly emerging. The strains detected in the UK, as well as delta and omicron strains, seemingly emerged out of nowhere and spread at lightning speed. All these strains differ significantly from the original Wuhan strain and from each other by multiple mutations. However, scientists were unable to detect any intermediate or transitional variants that would show a consistent accumulation of these changes.
"The evolution of a virus within a population can be likened to a journey across a vast terrain with ravines, valleys, and hills. The virus roams randomly through this landscape, dying quickly if it falls into a cavity, surviving longer in valleys, and thriving when on peaks," says the study co-author, Senior Researcher of the HSE International Laboratory of Statistical and Computational Genomics Alexey Neverov.
"The fitness landscape of the coronavirus is more like a vast expanse of water with scattered islands representing the virus variants which differ from each other by a specific set of mutations. To move from one island to another, one must travel by boat for a long while and avoid drowning. Science does not yet have a precise answer as to how the virus travels between these islands."
Having analyzed over three million genome sequences of different strains of SARS-CoV-2, the Russian researchers were able to identify particular sites on the surface protein of the coronavirus where amino acid substitutions occurred, differentiating the variants from both the original Wuhan strain and from one another.
Many of these sites appeared to be concordantly evolving, so that changes in amino acids at one site were rapidly followed by changes at another site. All the active and dangerous variants of the virus were distinguished from the previously prevalent variants by patterns of multiple substitutions.
"The process of moving from one island to another involves an accumulation of mutations. While the virus is 'afloat,' it is vulnerable and poorly adapted. Indeed, it can only reach another island if it has some kind of a boat. Regarding the coronavirus, individuals with long-term COVID-19 may potentially harbor an accumulation of variants which are poorly adapted for survival in the general population. However, over time, these variants can evolve into stronger forms that have the potential to spread widely and conquer the world," Neverov explains.
The study authors suggest an explanation as to why intermediate variants that differ from the original virus by only one or two substitutions may not be visible. It's possible that these 'weak' variants only become 'strong' when they gather the whole pattern of individually deleterious substitutions. As a result, predicting the emergence of a new highly adaptive strain is hard.
The statistical method employed by the study's authors is versatile and can be utilized to investigate the evolution of numerous other pathogens. In particular, this approach has been successfully applied to studying the evolution of influenza and tuberculosis.
More information: Alexey Dmitrievich Neverov et al, Coordinated evolution at amino acid sites of SARS-CoV-2 spike, eLife (2023). DOI: 10.7554/eLife.82516
Journal information: eLife
Provided by National Research University Higher School of Economics