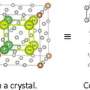
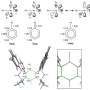
Structural determination of complex anion materials
 and crystal structure model (right) obtained by nuclear magnetic resonance experiments of the composite anion material Pb2Ti4O9F2. The two peaks appearing in the waveform demonstrate the two types of atomic positions are selectively occupied when some of the oxygen in the original solid is replaced by fluorine. Using density functional theory-based calculations, the researchers explained the preference of fluorine occupation on these sites. Credit: Ryo Maezono from JAIST")
Solid-state materials are widely used in semiconductors, phosphors, and batteries, and have become an indispensable part of modern life. Substitution of elements in these complex composite materials is a popular technique to achieve desired material properties. Various properties can be achieved by partially replacing oxygen in the oxide solid with another element such as fluorine (composite anion technology).
However, to tune material properties by substitution, it is important to know the sites in the material where the element is substituted. If the substitution site is near the highly reactive site in the solid, it favors the reaction that develops a certain material property.
To this end, a team of researchers led by Professor Ryo Maezono, from the Japan Advanced Institute of Science and Technology, developed an analytical tool to investigate the ordering of fluorine in lead titanium oxyfluoride, a complex composite material.
In a recent study published in Dalton Transactions on September 23, 2022, the researchers report developing an interdisciplinary method for clarifying the positions of substitution atoms in complex composite materials. To overcome the limitation of determining the sites of substitution by experimental techniques alone, the team employed advanced computational simulations. As Professor Maezono explains, "We have established a method for clarifying the positions of substitution atoms in solid materials, which cannot be clarified only by experiments, by computer simulation."
The researchers used a simulation called "first-principles calculation" (density functional theory) to analyze the experimental results and determine the element substitution positions in the composite anion material. The team succeeded in identifying the element substitution positions for composite anion materials where some of the oxygen atoms are replaced by fluorine.
Simulations were performed using first-principles calculations for a crystal structure model with various element substitution positions, and each energy value was compared. The results show that the substitution position that gives the lowest energy value is the likely position for substitution. Further simulations were performed using the crystal structure model with the substitution positions determined in this way and results consistent with the data observed in various experiments were obtained.
This analysis shows that in lead titanium oxyfluoride, the fluorine atoms predominantly occupy two of the six available inequivalent sites in a ratio of 73:27. The researchers explained the preference of fluorine occupation on these sites using density functional theory-based calculations that matched the experimentally observed occupation ratio. They further explained that the lead atom valence electrons could potentially determine the majority and minority fluorine occupation sites.
Using supercomputing facilities, faster simulations are now able to determine which substitution position has the least discrepancy with the experiment. This approach complements experimental observations to reveal the mechanism of anion ordering in complex materials. This result makes it possible to provide a powerful analytical tool in the field of material development, in which atomic-level substitution is performed on solid materials and their properties are tuned.
Professor Maezono concludes, "The methodology developed in this work can accelerate the development of mixed-anion materials. Mixed-anion technique could realize better materials than the conventional mono-anion materials in semiconductor spintronics industries."
More information: Kengo Oka et al, Anionic ordering in Pb2Ti4O9F2 revisited by nuclear magnetic resonance and density functional theory, Dalton Transactions (2022). DOI: 10.1039/D2DT00839D
Journal information: Dalton Transactions
Provided by Japan Advanced Institute of Science and Technology