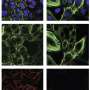
New strategy to precisely target subtypes of key protein
 binding and inhibiting Cyclophilin D protein. Credit: Aziz Rangwala")
Cyclosporine is one of the most common and effective immunosuppressant drugs used to treat chronic diseases like arthritis and psoriasis, but it comes with a risk of serious side effects. Scientists think that may be because the drug broadly targets cyclophilins, a family of 17 regulatory proteins that play different roles in promoting cellular health. Although each individual cyclophilin subtype has a unique role, many current immunosuppressive drugs target the entire family, meaning that important unknown pathways may be accidentally turned off or otherwise altered.
The problem is complicated by the fact that the active site where molecules bind is almost identical across all 17 cyclophilins, making it tough for drugmakers to target specific subtypes. In a paper published today in Nature Chemical Biology, scientists in the lab of Broad Institute Core Member David Liu, who is also the director of the Merkin Institute of Transformative Technologies in Healthcare at Broad, in collaboration with the labs of Markus Seeliger at SUNY Stony Brook and Institute Member Vamsi Mootha at Massachusetts General Hospital have proposed a new solution.
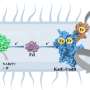
Rather than targeting the active site of cyclophilin proteins, researchers in Liu's lab describe a process that finds compounds that bind to the "exo site," a small pocket next to the active site that varies in size and shape across different cyclophilins. Using isolated proteins in a test tube, the team discovered several compounds that exclusively bind and inhibit Cyclophilin D (CypD), a protein involved in the opening and closing of mitochondrial pores. They also applied similar principles to discover unique, selective inhibitors for Cyclophilin E (CypE). The authors say that their study lays the groundwork for scientists to develop additional subtype-selective cyclophilin inhibitors, some of which may be useful as tools for biology or as leads for therapeutic development.
"It's a new binding mode that takes advantage of a pocket that people haven't fully explored yet," lead author Alex Peterson, now a postdoctoral fellow at the Scripps Research Institute, who led the project as a graduate student in Liu's lab, said. "It's kind of a blueprint for how people can design selective cyclophilin inhibitors going forward."
Leveraging new, and old, technologies
CypD regulates the mitochondrial permeability transition pore (mPTP), small pores located on the inner surface of mitochondria (famously known as the powerhouse of the cell). When CypD detects oxidative stress or high calcium levels, it rushes to open the mPTP, allowing water and other ions to rush in and out of the mitochondria.
This opening of the mitochondrial floodgates can become a problem with diseases like ischemia reperfusion injury, diabetes, neurodegenerative disorders, liver diseases, and more. Since these conditions can cause abnormally high levels of oxidative stress, CypD holds the mitochondrial pores open for longer than usual, causing mitochondrial dysfunction, rupture, and cell death. It's been thought that drugs that slow down and inhibit CypD's reaction to high oxidative stress might be used to treat a host of diseases.
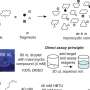
To track down compounds that exclusively bind to CypD, the team turned to DNA-encoded small-molecule libraries, a technology developed over twenty years ago as one of the first project's in Liu's then-new lab. Researchers can use the libraries, which are filled with hundreds of thousands of synthetic compounds attached to unique DNA barcodes, to scan for molecules that bind to desired proteins. By mixing isolated CypD proteins and a collection of 256,000 unique DNA-encoded compounds in a test tube, the team identified hundreds of promising compounds.
Most of the initial compounds still bound in and around the active site, inhibiting multiple cyclophilin subtypes, so the team gradually made small chemical changes to their compounds to make them unique to CypD. Once they discovered that the exo site was the key to developing subtype specific inhibitors, they were able to design a pair of compounds that potently inhibit CypD while minimally affecting other cyclophilins. X-ray co-crystal structures of the CypD protein and the inhibitors during development gave the team a behind-the-scenes look at the precise location where their molecules were binding.
The researchers then treated isolated mitochondria with their two leading compounds and observed that they were effective in slowing down CypD's opening of mitochondrial pores. The mirror images of their compounds, which do not inhibit CypD, did not show activity in mitochondria. To prove that their success wasn't an isolated incident, they repeated the strategy again for CypE, a cyclophilin responsible for regulating mRNA processing. Once again, they developed a compound that exclusively targeted it and left the remaining 16 cyclophilins unphased.
The team hopes their findings can ultimately help chemical biologists and drugmakers build better and more specific cyclophilin targeting drugs. They even gave future scientists a leg up—because the CypD-targeting compounds struggle to enter human cells on their own, the team tweaked them by adding ester derivatives that effectively bypass the plasma membrane and deliver into mitochondria.
"Our team's work eventually allowed us to conquer this long standing problem: how do you selectively inhibit just one cyclophilin subtype out of 17?" said Liu, who is also a Howard Hughes Medical Institute investigator. "In the future, molecules that come from using our strategy will, I hope, prove to be useful both for basic science and potentially for therapeutics."
More information: Alexander A. Peterson et al, Discovery and molecular basis of subtype-selective cyclophilin inhibitors, Nature Chemical Biology (2022). DOI: 10.1038/s41589-022-01116-1
Journal information: Nature Chemical Biology
Provided by Broad Institute of MIT and Harvard