Researchers develop new method for analysing genetic admixture of populations

Researchers of the HSE International Laboratory of Statistical and Computational Genomics together with their international colleagues have proposed a new statistical method for analyzing population admixture that makes it possible to determine the time and number of migration waves more accurately. The history of Colombians and Mexicans (descendants of Native Americans, Spaniards and Africans) features two episodes of admixture that occurred about 350 and 200 years ago for Mexicans and 400 and 100 years ago for Colombians. The results were published in PLOS Genetics.
When Francis Crick and James Watson deciphered the structure of DNA in 1953, they declared that they had 'found the secret of life." Indeed, all life on Earth is reproduced by constant cell division and copying of its genetic material. DNA is passed down from generation to generation, and the human genome is a mosaic of genetic fragments of our ancestors from different times. To understand the origins of the genetic diversity of modern humans, it is necessary to study the history of populations: where our ancestors lived, when and where they migrated, when and how they mixed.
The history of population admixture can be uncovered by analyzing the connections between human genetic variants. Our genome has genetic material from our father and mother; then we pass on new combinations of genetic variants, a mosaic made up of the genomes of our parents, to our descendants. This phenomenon is called recombination.
For example, a Spanish mother and a Native American father will have a child with one Spanish and one American set of chromosomes. Their child in turn will pass on a set of chromosomes that includes a combination of sections of Spanish and American origin to their descendants (the second set of chromosomes will be inherited from the other parent). The origin of these sections can be determined by the sequences of genetic variants typical for a particular population. In each new generation, recombination will mix sections of different origins more and more, breaking up these typical genetic sequences. Over time, they disintegrate, finally mixing with each other.
Thus, by calculating the correlation between genetic variants on different parts of chromosomes and analyzing the strength of their connections, we can say how many generations ago population admixture occurred.
Earlier methods of analyzing the genetic admixture of populations were capable of estimating the time of the last admixture event. The algorithm was based on the analysis of the connection strength between pairs of genetic variants. Researchers from the HSE International Laboratory of Statistical and Computational Genomics and their international colleagues proposed analyzing triple variants. This statistical method makes it possible to model more complex scenarios of population admixture, for example, to identify two episodes of admixture and determine how many generations ago they occurred.
"Let's imagine that ships with European settlers land on the shores of America for the first time. Europeans start exploring new territories and mixing with the indigenous population of America. However, after a few generations, more ships with Europeans arrive in America. Our method allows us to see that there were two waves of resettlement, two episodes of admixture in different time periods," explains Mikhail Shishkin, co-author of the article, research assistant of the laboratory and MIEM student.
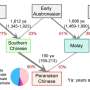
As an example, the paper's authors analyzed genetic samples of the population of Colombians and Mexicans from the genetic database of 1000 Genomes. Both populations appeared as a result of admixture of Native Americans, Spaniards and Africans. The results showed that the history of both populations featured two waves of admixture, which occurred 13 and 8 generations (350 and 200 years) ago for Mexicans and 15 and 4 generations (400 and 100 years) ago for Colombians.
"Our method requires large amounts of data —if earlier algorithms required dozens of samples, then we need hundreds. And today we can get them. In our case, we used the genetic database of the 1000 Genomes project. Over the past 10 years, the possibilities of genome sequencing and data processing have expanded significantly, so that the number of available samples no longer limits us," says Vladimir Shchur, Head of the HSE University International Laboratory of Statistical and Computational Genomics.
More information: Mason Liang et al, Estimating the timing of multiple admixture events using 3-locus linkage disequilibrium, PLOS Genetics (2022). DOI: 10.1371/journal.pgen.1010281
Journal information: PLoS Genetics
Provided by National Research University Higher School of Economics