An algorithm to make CRISPR gene editing more precise

Researchers from Aarhus University and University of Copenhagen have developed a new method, which makes CRISPR gene editing more precise than conventional methods. The method selects the molecules best suited for helping the CRISPR-Cas9 protein with high-precision editing at the correct location in our DNA, the researchers explain.
It eventually became a Nobel prize-winning revolution when researchers first engineered CRISPR as a gene editing technology for bacterial, plant, animal and human cells. The potential of the technology is great and span from curing genetically disposed diseases to applications in agricultural and industrial biotechnology, but there are challenges.
One such challenge consists of selecting a so-called gRNA molecule which should be designed to guide the Cas9 protein to the right location in the DNA where it will make a cut in relation to the gene editing.
"Typically, there are multiple possible gRNAs and they are not all equally efficient. Therefore, the challenge is to select the few that work with high efficiency and that is precisely what our new method does," says Yonglun Luo, Associate Professor Department of Biomedicine at Aarhus University.
The new method is developed from the researchers' new data and implementation of an algorithm, which gives a prediction on what gRNAs that work most efficiently.
"By combining our own data with publicly available data and including knowledge on the molecular interactions between gRNA, DNA and the CRISPR-Cas9 protein, we have succeeded in developing a better method," says Jan Gorodkin, professor at the Department of Veterinary and Animal Sciences at the University of Copenhagen.
Data, deep learning molecular interactions
Gorodkin's research group with Giulia Corsi and Christian Anthon have collaborated with Luo's research group in order to achieve the new results. The experimental part of the study was conducted by Luo's group while Gorodkin's group spearheaded the computer modeling.
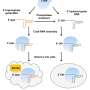
"In our study, we have quantified the efficiency of gRNA molecules for more than 10.000 different sites. The work was achieved using a massive, high throughput library-based method, which would not be possible with traditional methods," says Luo.
The researchers took their starting point concerning data generation in the concept of having a virus express gRNA and a synthetic target site in one cell at a time. The synthetic target sites have exactly the same DNA sequences as the corresponding target sites in the genome. Thus, these synthetic target sites are used as so-call surrogate target sites to capture the CRISPR-Cas9 editing efficiency. Together with colleagues from Lars Bolund Institute of Regenerative Medicine in BGI-Research and Harvard Medical School, they generated high quality CRISPR-Cas9 activity for over 10,000 gRNAs.
With this dataset of gRNAs with known efficiencies from low to high, the researchers were able to construct a model that could predict efficiencies of gRNAs which has not been seen before.
"In order to train an algorithm to become precise, one has to have a large dataset. With our library of viruses, we have obtained data that constitutes the perfect starting point for training our deep learning algorithm to predict the efficiency of gRNAs for gene editing. Our new method is more precise than other methods currently available," says Gorodkin.
More information: Xi Xiang et al, Enhancing CRISPR-Cas9 gRNA efficiency prediction by data integration and deep learning, Nature Communications (2021). DOI: 10.1038/s41467-021-23576-0
Journal information: Nature Communications
Provided by University of Copenhagen