Cellular exclusion of mitochondria protects cells from damage

Mitochondria are cellular organelles that generate most of the energy cells need to function, and thus play an important role in maintaining cell health. In a new study, researchers from Osaka University discovered a novel mechanism by which cells turn over mitochondria and contribute to cellular maintenance. They further showed how this novel pathway is involved in the development of Parkinson's disease (PD).
Most cells in the body require mitochondria to function and survive. Various disease processes and toxic agents can harm cells by damaging mitochondria, and failure to eliminate damaged mitochondria can cause neurodegenerative diseases such as PD. Unsurprisingly, cells have a quality control system that ensures the health and function of mitochondria within cells. For a long time, it was thought that this control system would consist of mitophagy only, a process by which cells degrade damaged mitochondria and recycle their individual components to then build new mitochondria. While recent studies have shown that cells in addition have the ability to transfer mitochondria out, this process remains incompletely understood.
"The release of mitochondria into the extracellular space is a fascinating topic, but we know very little about it," says co-author Tatsusada Okuno. "The goal of our study was to investigate to which extent cells transfer mitochondria out in response to mitochondrial stress."
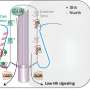
To achieve their goal, the researchers generated a cell line in which mitochondria are fluorescently labeled and thus can be investigated under a fluorescence microscope. By performing time-lapse imaging over a prolonged period of time, the researchers were able to watch the release of mitochondria in real-time. Intriguingly, they were able to show by correlative light-electron microscopy, a technique that allows cellular structures to be examined at ultra-high magnification, that mitochondria were mostly released in free form and were rarely enclosed in vesicles.
 and electron microscopic image (right: III, IV) of mitochondria released from PC12 cells. Credit: Osaka University")
The researchers then asked what the role of mitochondrial release might be in PD. By studying cells deficient in parkin, the protein responsible for the hereditary form of PD, they were able to show markedly increased mitochondrial release in the absence of the protein. Likewise, the blood of parkin-deficient mice contained higher levels of mitochondrial proteins, further demonstrating how the absence of parkin facilitates extracellular mitochondrial release. Most importantly, however, the researchers were able to show that cerebrospinal fluids of PD patients with a mutation in parkin that renders the protein dysfunctional contained higher levels of mitochondria, demonstrating the significance of this novel mitochondrial quality control system in the development of PD in humans.
 is markedly increased in the cerebrospinal fluid of Park2 patients. Credit: Osaka University")
"These are striking results that demonstrate a novel mechanism for clearing defective mitochondria and an important role for mitochondrial homeostasis in the onset of Parkinson's disease," says corresponding author Hideki Mochizuki. "These findings may lead to the development of new biomarkers and therapeutics."
More information: Chi-Jing Choong et al. Alternative mitochondrial quality control mediated by extracellular release, Autophagy (2020). DOI: 10.1080/15548627.2020.1848130
Provided by Osaka University