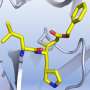
Gold standard force fields help identify promising peptides to disrupt COVID-19
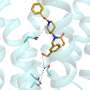
, University of Utah researchers rapidly generated molecular models of compounds relevant for COVID-19, in collaboration with medicinal chemists. Credit: Cheatham Lab")
Drug discovery is arduous, expensive, and often prone to failure, but computer-aided drug design and simulation can speed up and improve the development of treatments.
Thomas Cheatham—a professor of medicinal chemistry and director of the Center for High Performance Computing at the University of Utah—and Rodrigo Galindo—a research professor in his group—use powerful supercomputers to predict the characteristics of novel drugs.
Recently, they have been using Frontera, the fastest supercomputers at any university in the world, and Longhorn, an IBM/NVIDIA system at the Texas Advanced Computing Center (TACC), to rapidly generate molecular models of compounds relevant for COVID-19, in collaboration with medicinal chemists.
How molecules bend, bind or react with proteins in a cellular environment depends strongly on their molecular force fields (not in the Star Trek sense of the term, but rather the potential energy of a system of interacting atoms). However, force fields are extremely challenging to predict accurately.
One of the leading tools used to simulate force fields and their impacts on binding is Amber (Assisted Model Building with Energy Refinement), a molecular simulation suite of software and force fields, for which Cheatham is one of the main developers. Known as one of the first community codes, Amber has also been evolving since 1978; its simulations and force fields get more accurate with each passing year.
For relatively simple structures, Amber can match experimental results to within less than half an angstrom (Å)—one hundred-millionth of a centimeter, 10-10 meter.
Cheatham and Galindo apply Amber to biomolecular simulations with applications in medicine. "The goal is to understand the structure, function, dynamics, and energetics of biomolecular systems in their native environment, with water and other ligands."
They were in the midst of two biomolecular studies—exploring a promising class of cancer-fighting copper-compounds and investigating how experimental modifications to the backbone of DNA can resist degradation inside the body—when the COVID-19 pandemic struck.
Cheatham and Galindo swiftly turned their attention to discovering drugs that could disrupt the coronavirus.
In 2015, supported by an NSF RAPID grant, Cheatham had developed an approach to rapidly identify molecules to stop the Ebola virus. The workflow, based on known crystal structures, uses the Rosetta software suite to select promising amino acid side chains on a fixed peptide backbone template and then performs molecular dynamics simulations with Amber to optimize the structures, which are then ranked based on free energetics estimates.
Galindo rapidly generated more than 2,000 molecular models of compounds relevant for COVID-19 using this approach on the Longhorn and Frontera supercomputers at TACC. The success of the effort led Cheatham and Galindo to apply for, and obtain, 2.7 million node hours on Blue Waters, another GPU-based system located at the National Center for Supercomputing Applications (NCSA), to continue their efforts.
The award was granted through the COVID-19 HPC Consortium—a public-private consortium working to match researchers with resources to accelerate virus research.
They are investigating the crystal structure of the COVID-19 main protease—an enzyme which breaks down proteins and peptides—in complex with a peptide inhibitor N3 that was recently published. They plan to apply the Ebola peptide design workflow to this system to explore the ability of the COVID-19 main protease to break down a series of similar, easy to detect probes, which have already been designed. This will serve as the basis for protease assays which test the efficacy of the inhibitors.
The peptide leads will later be transformed into circular modified peptides by the Schmidt lab in the Medical Chemistry department at the University of Utah, which has considerable experience in this area. Circular peptides are promising scaffolds for biopharmaceuticals that can be applied to the inhibition of protein-protein interactions, and protease inhibition.
Cheatham and his team were among the first to use the Longhorn and Frontera systems for COVID-19 research. That access is helping to fast-track treatment options.
"Our hope is that we find a new peptide inhibitor that can be experimentally verified in the next couple of weeks," Cheatham said. "And then, we will engage in further design to make the peptide cyclic to make it more stable as a potential drug. The hope is we can, in the next few months, find and experimentally verify a better peptide inhibitor for the COVID main protease."
Provided by University of Texas at Austin