Modeling a model nanoparticle
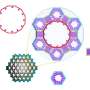
 The BE of CO on various sites of Au NPs as a function of CElocal: 172-atom cube (rectangles), 147-atom icosahedron (hexagons), and 147-atom cuboctahedron (rhombus). Heat map of different sites on the NPs with respect to their BE of CO (B to D) and to their CElocal (E to G). The color scheme follows the range of strongest CO binding to weakest CElocal (violet) and of weakest binding to strongest CElocal (red). Credit: Science Advances (2019). DOI: 10.1126/sciadv.aax5101")
Metal nanoparticles have a wide range of applications, from medicine to catalysis, from energy to the environment. But the fundamentals of adsorption—the process allowing molecules to bind as a layer to a solid surface—in relation to the nanoparticle's characteristics were yet to be discovered.
New research from the University of Pittsburgh Swanson School of Engineering introduces the first universal adsorption model that accounts for detailed nanoparticle structural characteristics, metal composition and different adsorbates, making it possible to not only predict adsorption behavior on any metal nanoparticles but screen their stability, as well.
The research combines computational chemistry modeling with machine learning to fit a large number of data and accurately predict adsorption trends on nanoparticles that have not previously been seen. By connecting adsorption with the stability of nanoparticles, nanoparticles can now be optimized in terms of their synthetic accessibility and application property behavior. This improvement will significantly accelerate nanomaterials design and avoid trial and error experimentation in the lab.
"This model has the potential to impact diverse areas of nanotechnology with applications in catalysis, sensors, separations and even drug delivery," says Giannis (Yanni) Mpourmpakis, the Swanson School's Bicentennial Alumni Faculty Fellow and associate professor of chemical and petroleum engineering, whose CANELa lab conducted the research. "Our lab, as well as other groups, have performed prior computational studies that describe adsorption on metals, but this is the first universal model that accounts for nanoparticle size, shape, metal composition and type of adsorbate. It's also the first model that directly connects an application property, such as adsorption and catalysis, with the stability of the nanoparticles."
More information: James Dean et al, Unfolding adsorption on metal nanoparticles: Connecting stability with catalysis, Science Advances (2019). DOI: 10.1126/sciadv.aax5101
Journal information: Science Advances
Provided by University of Pittsburgh