Hook-on drugs: New delivery strategy for K-Ras disruption

"The strategy was to design the drug to be able to hook into the hole of the FTase and GGTase I, otherwise the surface of the proteins are too large and slippery," Dr. Junko Ohkanda of Shinshu University explains her strategy behind her paper chosen by Chemistry—A European Journal as a "Hot Paper."
Pharmaceutical companies around the world have been trying to concoct an effective drug to target K-Ras proteins for the last 20 to 30 years. When K-Ras proteins mutate, they cause the multiply switch to remain perpetually on, becoming an aggressive and untreatable form of cancer. In 90 to 100% of the difficult lung and pancreatic cancers, K-Ras is said to play a role. 30% of all cancers is said to have some form of Ras mutation.
Scientists have had trouble designing a drug to infiltrate K-Ras due to a lack of interactive pockets. A new strategy was devised to attack the FTase, an important enzyme in the lipid modification of K-Ras. Without FTase, the mutated K-Ras would be unable to multiply uncontrollably. Scientists have developed large numbers of FTase inhibitors, but found it difficult to inactivate K-Ras modification.
Even when the FTase was inhibited, K-Ras modifications were not stopped because GGTase I was also reacting with the K-Ras, despite its different reactive cavity. It was not understood why, until its mechanism was elucidated that FTase and GGTase I are both made of two protein parts, one of which is the same, with the exact same DNA.

Near the activated cavity FTase and GGTase I have the same cluster of acidic amino acids, like glutamic acid and aspartic acid, carrying a negative charge. When closely observing the K-Ras C-terminus, it had an interactive positive charge. Other Ras proteins do not have this positively charged area. Only K-Ras has this cluster of positive charges. This is why even if the FTase was inhibited, the K-Ras still reacted with the GGTase I, even though its cavity was different.
This is where Dr. Ohkanda had her moment of inspiration. In theory, the pocket of the enzyme and the cystine key attach and join together. But in this case the surfaces of the proteins, with the plus and the minus also interact. Even if the FTase is inhibited the K-Ras mistakenly interacted with the GGTase I. Dr. Ohkanda and her colleagues thought with one compound they could work two functions.
The strategy was to design a molecule to mimic the part of the K-Ras that acts on the active pocket and also the acidic surface. It goes without saying that the function of the drug must happen inside the cell. Large molecules that are useful in protein-protein interactions are often too large to go inside the cell. This is a problem that riddles many drug developers: delivery methods.
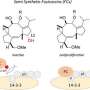
Dr. Ohkanda thought if she could rationally design the thiol on the end of the K-Ras to hook on to the active pocket of the FTase and GGTase I, the extended interactive positive charge portion could interact and penetrate the cell membrane. If the cysteine portion could hook on into the cavity, the connected interactive positive chain can be small and delivered strategically to the acidic surface of the enzymes.. It was difficult to minimize the size of the compound while increasing its stability and keeping its ability for chemical reactions. By using a peptidomimetic of the same length and same key, they were able to successfully penetrate the cell in vitro, disrupting the runaway K-Ras multiplication.
More studies are needed to increase the activity of the compound, test in vivo and to evaluate its toxicity long before the compound can be used as a treatment for cancers. Dr. Ohkanda continues to work with an international team of experts to elucidate the mechanism of action and their interactions to rationally design effective drugs to stop the multiplication of such cells.
More information: Mai Tsubamoto et al, A Guanidyl‐Based Bivalent Peptidomimetic Inhibits K‐Ras Prenylation and Association with c‐Raf, Chemistry – A European Journal (2019). DOI: 10.1002/chem.201903129
Journal information: Chemistry – A European Journal
Provided by Shinshu University