Researchers develop new method for deriving 3-D protein structures from lab-designed DNA sequences, mutant genes

Sequencing the first human genome was a herculean effort that took 13 years, hundreds of researchers around the globe and billions of dollars.
But recent advances in technology have transformed genome sequencing into a fairly mundane affair with millions of people having sequenced at least parts of their genomes using DNA collection kits available at drug stores.
Although these leaps in scientists' ability to perform genetic analyses have yielded untold insights into human heritage, disease and health, the precise meaning behind DNA sequences—how the order of the "letters" in each DNA strand instruct the body's proteins what to become and what to do—remains unclear.
Now, in a scientific first, scientists in the Blavatnik Institute at Harvard Medical School have shown it is possible to determine the 3-D structures of a gene by assessing the effects of lab-made genetic mutations on protein functions.
The team's findings, published June 17 in Nature Genetics, represent a significant step toward linking sequence data with its function in cells. The tool is freely available at https://github.com/debbiemarkslab/3D_from_DMS_Extended_Data .
The current study will be published in parallel with research from a team led by Jörn Schmiedel and Ben Lehner at the Barcelona Institute of Technology that independently arrived at similar results, employing the concept but using a different technique, thus affirming the utility of the approach.
The computational approach used in the study is known as deep-mutational scanning and involves the use of high-throughput sequencing to synthesize various genetic mutations and then determine the mutations' impact on protein function.
By contrast, previous efforts—including work by researchers in the current study—relied on machine learning to glean such 3-D structures from naturally occurring, rather than lab-made, DNA samples.
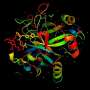
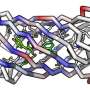
In the current study, researchers identified functional interactions within DNA sequences containing instructions for four different proteins and one RNA. From these, the researchers constructed 3-D structures of the proteins—a spatial configuration that can lend valuable clues about the work these proteins perform in cells.
"We live in a three-dimensional world where structure determines function," said study senior investigator Debora Marks, an associate professor of systems biology at Harvard Medical School, who led the team with HMS post-doctoral researcher Kelly Brock and doctoral student Nathan Rollins of Harvard University.
"Understanding what shapes and conformations proteins take inside cells can help us predict their function and the effects that variations in these structures can have on cell function or malfunction."
Such insight, Marks said, represents a marked advance toward a better understanding of individual protein variations in disease and health and can inform the development of precision drugs that target specific parts of the proteins. Each protein in the human body is made of a string of combinations of 20 different amino acids, Marks explained. The amino-acid makeup of the protein is important, but how these amino acids fold, interlace and relate to each other three-dimensionally is just as critical in determining protein function and dysfunction.
Researchers have long relied on methods such as X-ray crystallography, cryogenic electron microscopy (cryo-EM) or nuclear magnetic resonance imaging (NMRI) to determine protein structures. However, these methods can be time-consuming and require expensive, highly specialized equipment. Some proteins, such as those bound to membranes or those that tend to aggregate, or clump—such as amyloids in the brain—aren't amenable to these visualization techniques at all.
Searching for a better way, Marks and her colleagues from Harvard Medical School, the Dana-Farber Cancer Institute and the Broad Institute turned to mutational libraries—synthetic DNA sequences developed by other researchers in which the pattern of DNA was changed to alter individual amino acids.
The team was interested specifically in libraries containing simultaneous mutations in separate amino acids within the same sequence. They sought mutation pairs that affected protein function and fitness interdependently of one another, a biologic effect known as epistasis. The strongest instances of epistasis, the investigators reasoned, should be mediated by direct interactions between the amino acid partners in 3-D. Strong epistasis in a scan with enough mutation pairs should thus reveal sufficient 3-D interactions to glean and build the full 3-D structure.
The team used proteins derived from humans, yeast and rice, including a two-protein complex, as well as a ribozyme, a piece of RNA with enzyme-like function. Each of these molecules was well-studied and had existing structures derived through other means, which allowed the team to validate the precision of their final predictions.
The researchers fed information from these libraries into a computer program, which they used to generate the 3-D structures of the molecules. Surprisingly, data from these epistatic mutations were enough to generate structures that closely mimicked those derived from the established methods, with variations in physical positions as small as 1.8 angstroms.
Although this method is amenable for use in small proteins, Marks noted, larger proteins present a greater challenge. For example, a protein composed of 300 amino acids would have 16 million possible mutation pair sequences. While sequence synthesis is becoming vastly more efficient and may soon shoot past this limitation, it remains difficult to create libraries of that size.
However, additional work showed that running this full dataset isn't necessary; running just a fraction of the possible mutational combinations produced accurate 3-D structures. The team showed that libraries as small as one-twentieth of the original size could get the job done when applying simple rules for selecting mutants to synthesize. As the method evolves, strategies for even more efficient libraries will likely emerge and perhaps circumvent the synthesis challenge altogether.
Marks and her colleagues noted that this method can be expanded far beyond the types of molecules that they studied here. For example, she said, their work is already spurring collaborations with other groups interested in learning the structures of proteins that assume different shapes to perform different functions. In addition, the approach might also be used to study other types of RNA, a broad class of molecules that are heavily involved in human disease but have few solved structures.
"This approach doesn't replace X-ray crystallography or nuclear magnetic resonance as ways to derive 3-D structure," Marks said. "But it's another tool in our toolbox for better understanding these structures and learning how they work."
More information: Inferring protein 3D structure from deep mutation scans, Nature Genetics (2019). DOI: 10.1038/s41588-019-0432-9 , www.nature.com/articles/s41588-019-0432-9
Journal information: Nature Genetics
Provided by Harvard Medical School