I build mathematical programs that could discover the drugs of the future
")
Discovering and designing a new drug is one of the most challenging tasks in the biological sciences. It takes over 10 years and about US$2.6 billion to bring an average drug to market.
The essential idea behind making a new drug is to create a molecule that binds to a target, such as a protein or DNA, so as to activate or inhibit its biological function. That, in turn, should lead to a therapeutic benefit to the patient.
Ideally, the drug should be highly selective, meaning that it binds strongly to the target molecule without affecting any other similar molecules and that it gets along with the human metabolic system. There are many steps before the drug can come to market, including toxicity tests, three phases of clinical trials and measurements to determine the appropriate dose.
While many people were surprised by the power of artificial intelligence when AlphaGo beat human experts at Go for the first time a few years ago, it was not clear whether AI could tackle scientific challenges.
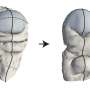
But, last December, Google's DeepMind won the Critical Assessment of Structure Prediction competition using its latest artificial intelligence system, AlphaFold. This competition recognizes the technology that can best construct a protein's 3-D structure from biological sequences. AlphaFold was able to win 25 out of 43 contests with high accuracy. By comparison, the next best team won just three.
AlphaFold's dominant win ushers in a new era of scientific research. Scientists like myself are excited about what the future may hold for biomedicine. AI will make discovering new drugs significantly faster and cheaper. This could be particularly beneficial to patients with rare medical ailments, for whom drug discovery is currently not profitable or for those whose medical ailments currently can't be effectively treated with drugs, such as Alzheimer's disease.
Automated drug design
However, drug design is much more complex than protein folding prediction.
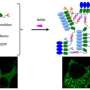
Generative adversarial networks, a cutting-edge type of artificial intelligence program, could be a prototype for automated drug discovery. One part of the program generates potential molecules that could be used in drugs, while the other weeds out unqualified candidates based on existing database information and ideal drug requirements.
The generator learns from the mistakes caught by the discriminator until the discriminator is satisfied.

One of the major obstacles for automated drug design is the extreme size of the problem. A typical human protein and its binding drug have many, many possible configurations. Even if a computer enumerates one possible configuration per second, it would still take longer than the universe has existed to reach the right configuration.
Even all the world's computers put together don't have enough power to design drug automatically.
Untangling the molecules
In my lab, we tackle these challenges mathematically. Our work focuses on reducing the geometric complexity of individual proteins for computers. Each protein has a unique shape that affects what molecules it might bind to. We have introduced three mathematical methods that greatly increase the computer's ability to represent new drug molecules and how they might interact with proteins in the human body.
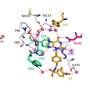
For example, the first method utilizes differential geometry, a technique used by Einstein to formulate his general theory of relativity that explained space, time and the motions of stars. We use it to describe the contact surfaces and interactions of proteins and drugs. That makes it easier for the computer to assess how the protein would react to a given drug molecule.
Another method is called algebraic topology, which provides a high-level abstraction of protein-drug interactions so that a computer can easily grasp the difference between a suitable drug and a decoy. The other method, graph theory, examines the wide variety of ways a protein might interact with a drug, such as hydrogen bonding, electrostatic interactions or reactions to water. By studying all of these possible interactions, we can look out for anything that might be harmful to human health.
The impact of our mathematical approaches is best related through D3R Grand Challenges, an annual worldwide competition where entrants pit their tools against prescribed drug posing and binding problems. We won 14 percent of contests in Grand Challenge 2 in 2016, improving this to a 38 percent win rate in Grand Challenge 3 in 2017. The second winner had a winning rate of 19 percent.
In the latest Grand Challenge 4, preliminary results indicate that we might have the best predictions in 50 percent of contests. Our approach is tens of thousands of times faster than traditional methods based on molecular simulations.
Stimulated by this success, we have entered into partnerships with Pfizer to improve mathematical AI tools for virtual screening and with Bristol-Myers Squibb to predict drug efficacy and optimize doses.
Integrating advanced mathematics and AI provides a powerful way to search for new drugs. It might be that, just a decade down the road, AI will become a leading force for new drug discovery, leading to less expensive and more efficient drugs.
Provided by The Conversation
This article is republished from The Conversation under a Creative Commons license. Read the original article.![]()