How this researcher's risky idea could mean big things for regenerative medicine

Every cell in your body contains thousands of different proteins. These complicated molecules regulate chemical reactions, bind to invading bacteria or viruses, carry signals in and between cells, and much more. They are vital to your existence.
But determining which proteins are in a particular cell is difficult. The best processes scientists have developed can identify less than 100 of them.
Now Nikolai Slavov, a researcher at Northeastern, has devised a method to identify more than a thousand proteins in an individual cell and estimate their abundance.
The ability to determine which proteins are in a cell has implications across the fields of medicine and biology. Recent developments have greatly expanded our understanding of the number of cell types in the human body. Identifying the proteins within those newly-discovered cells could explain their functions within our body and inspire new medical treatments. Studying the proteins within a cancerous cell could lead to a better understanding of how that cancer spreads.
It could also help scientists understand the signals that trigger stem cells, which have the potential to be any type of cell, to turn into a specific cell, such as a red blood cell or a muscle cell. If scientists can understand those signals, and what stem cells do when they receive them, then they might be able to instruct stem cells to become specific types of cells. This is the basis for regenerative medicine, treatments that may someday help us regrow damaged cells, tissues, and even organs.
"If we know what signals are active in cells that become, let's say beta cells in the pancreas," said Slavov, who is an assistant professor of bioengineering, "we can provide those signals to other cells that we want to instruct to become beta cells."
But identifying these proteins is no easy task. The genes in a single cell could produce on the order of 10,000 or 20,000 different proteins, Slavov said. And after they are made, proteins may go through different chemical interactions that tack on extra molecules and twist them into new shapes, creating completely different structures.
"If you take those into account, then there are hundreds of thousands, if not millions of proteins," Slavov said. "We haven't ever measured all of them."
A long shot
Experts in the field of proteomics, the large-scale study of proteins, thought it would be impossible to measure thousands of proteins within a single cell, Slavov said. Existing techniques weren't specific enough or required a much larger sample size than the contents of an individual cell.
"People thought that we were very far away from having the sensitivity to analyze material from a single mammalian cell," Slavov said. "I did not even pitch this when I started my lab."
But in the fall of 2015, as he was working on other projects in his new lab at Northeastern, Slavov began developing a process that could measure more proteins in a cell than ever before.
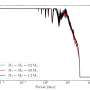
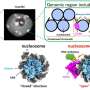
Slavov's method relies on mass spectrometry, a way of weighing molecules and their fragments. This has reliably been used to quantify proteins in the past, but typically requires a much larger sample size.
Slavov has figured out a way around that. After opening a cell and breaking the proteins up into smaller pieces called peptides, he tags them with molecules that serve as an identifiable barcode. Then he can mix in peptides from several hundred other cells, tagged with a different barcode, to round out the sample.
This makes the sample large enough to process and increases the likelihood that any peptides lost in preparing the sample will be from the unimportant filler material. It also makes it easier to identify the rarer peptides from the target cell. If the amount of a certain peptide is too low, the machine doesn't have enough information to tell what it is. Dumping in extra peptides, while still making them easy to separate by their barcodes, helps the more scarce molecules to register.
"This was the highest risk, highest reward idea that I had," Slavov said.
A preprint of the paper describing the work, which was recently published in the journal Genome Biology, was well-received by members of the proteomics community.
He tested this method with the help of Bogdan Budnik, who is the director of proteomics at the Harvard University Mass Spectrometry and Proteomics Resource Laboratory, and Ezra Levy and Guillaume Harmange, two Northeastern undergraduates doing co-ops in his lab. The students are listed as second and third authors on the Genome Biology paper.
Slavov said the undergraduates have been instrumental in getting the project going.
"I initially was reluctant to start this project with an undergraduate," he said, "But Ezra, a first- year undergraduate at the time, expressed very strong interest in the project and he certainly rose to the challenge."
Next steps
Identifying and quantifying the proteins in one cell is a good start. But Slavov wants to do much more.
"We are not interested in measuring just a single cell," Slavov said. "We want to measure tens of thousands of single cells."
If Slavov can tag the peptides from each new cell with a unique barcode, he can run them simultaneously. Right now he can analyze nine cells at a time, but is working with several colleagues to developing more barcodes. His lab has also recently released a preprint of a new paper improving on the method.
"This was never about a single method that was going to stay static," Slavov said. "This paper is much more of a proof of principle that opens the door to a lot of further development."
Slavov is already using this technique with physicians from Massachusetts General Hospital and Harvard Medical School. They are investigating the proteins in immune cells that are inadvertently helping the growth of certain types of cancer. But Slavov is particularly excited about the possibility of improving the scientific understanding of cell development and using that to direct stem cells.
"There is a whole community that is coalescing around those ideas," Slavov said. "Now that we have this proof of principle, I expect to see a lot of progress."
"This was an incredibly risky project that worked out great," he said.
More information: Bogdan Budnik et al. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation, Genome Biology (2018). DOI: 10.1186/s13059-018-1547-5
Harrison Specht et al. Automated sample preparation for high-throughput single-cell proteomics, (2018). DOI: 10.1101/399774
Journal information: Genome Biology
Provided by Northeastern University