DNA accessibility, gene expression jointly profiled in thousands of cells

Scientists have now developed an assay that concurrently profiles both the epigenome and transcriptome of each of thousands of single cells.
The epigenome and transcriptome are part of the molecular biology that converts a genetic blueprint into tools and materials for living cells.
The genomes of different types of cells may be identical, while their epigenomes and transcriptomes are not. The epigenome consists of a set of marks that shape what each cell's genome will do, while the transcriptome is the set of copies of the instructions themselves. These encode the production of proteins. The flow of information from the inherited plan to the making of proteins is critical for forming and maintaining life.
Cells can access only certain portions of their chromatin-packaged, double-stranded genome during RNA transcription. Because this access varies among different cell types, chromatin accessibility is what helps determine the shape, function and variety of the diverse cells in a multi-cellular, living organism.
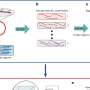
The researchers call their assay sci-CAR. Sci stands for single-cell combinatorial indexing, a means of studying large numbers of single cells at once. In a research report Aug. 30 in Science, the scientists describe how the new assay merges two other genomic assays into one protocol.
These assays, among their other features, incorporate unique barcodes for the nucleic acid contents of cells or of the cell nucleus, which contains the main control center for living cells. The scientists' method for labeling and sorting cells lets them link the messenger RNA and chromatin accessibility profiles of individual cells.
Most assays of what goes on genetically inside single cells, the scientists noted, can survey only one aspect of cellular biology. The ability to investigate several classes of molecules concurrently could uncover, for example, how certain genetic mechanisms are related and regulated.
It could also improve the usefulness of cell atlases of complex organisms, like those of the worm or mouse. Eventually, it could be helpful in compiling a human cell atlas.
The new method was developed by scientists at the Brotman Baty Institute for Precision Medicine in Seattle, University of Washington School of Medicine Department of Genome Sciences, Oregon Health Sciences University, Illumina, Inc., in California, Allen Discovery Center for Cell Lineage Tracing, and Howard Hughes Medical Institute.
The first author of the study is Junyue Cao, a graduate student in the Molecular and Cellular Biology program and in genome sciences at the University of Washington School of Medicine. The study was led by Jay Shendure and Cole Trapnell. Both are faculty in the Department of Genome Sciences at the UW School of Medicine and investigators at the Brotman Baty Institute, where Shendure is the director.
The researchers first tried their co-assay on more than 4,800 cells in a lung-cancer-derived cell culture model of cortisol response. In this model, the cells are treated with the corticosteroid dexamethasone. This synthetic steroid can activate the binding of thousands of locations on the genome and change the expression of hundreds of genes.
The scientists then examined the time course of dexamethasone's effects on gene expression, as well as dynamic changes that occurred in chromatin accessibility in the same cells.
In related work, the researchers sought to study the gene-control landscape that underlies the messenger RNA collections found in the different types of cells in the mammalian kidney.
In applying their co-assay to the nuclei from whole mouse kidneys, they recovered both transcriptome and chromatin accessibility profiles from 11,296 cells. They clustered their mouse kidney cells into 14 groups, and characterized cell-type specific epigenome landscapes and linked transcriptome features.
Based on the covariance between epigenome and transcriptome, the researchers also learned that they could draw links between distant genomic regulatory elements and their targeted genes to explain some of the differences in gene expression across various cell types.
Looking forward, there are clear advantages of a joint assay over assays that only profile either RNA transcription or DNA accessibility. One advantage of sci-CAR specifically is that this method could potentially be used to jointly assay millions of single cells at once.
Among its limitations is the sparseness of some of the chromatin accessibility data. The researchers suggested that this might be overcome in future experiments by optimizing some aspects of the current protocol.
The researchers hope to continue to combine additional co-assays so that molecular biologists could concurrently trace the flow of genetic information from DNA to RNA to specific proteins in each of the many single cells that can exist in complex living things.
More information: "Joint profiling of chromatin accessibility and gene expression in thousands of single cells" Science (2018). science.sciencemag.org/lookup/ … 1126/science.aau0730
Journal information: Science
Provided by University of Washington