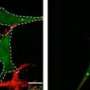
'Digital snapshots' reveal the protein landscape of mitochondrial quality control

Almost every cell in the human body, from beating heart cells to bacteria-gobbling immune cells, is powered by chemical energy produced by specialized organelles called mitochondria. The process of energy generation creates waste products that can be highly toxic, so, like real power plants, these organelles require tight quality control and oversight.
This life-or-death task ensures the survival of the cell and the organism to which it belongs. Malfunctions in a cell's ability to remove damaged mitochondria have been linked to conditions ranging from cancer to neurodegenerative disease—particularly Parkinson's, in which power-hungry neurons are highly vulnerable to the toxic effects of defective mitochondria.
In the April 19 issue of Molecular Cell, Harvard Medical School scientists report having developed a new technique to analyze, with unprecedented quantitative precision, how cells initiate the removal of defective mitochondria by the cell's autophagy, or "self-eating," system. The methodology allowed them to study, for the first time, this process in human neurons derived from stem cells.
By taking "digital snapshots" of the protein landscape as cells mark damaged mitochondria for autophagy, the research team generated the clearest picture to date of the dynamics of this process, including absolute measurements of protein modification reactions and their abundance and changes over time.
The results set the stage for detailed studies of the connections between mitochondrial damage, cell death and disease, and establish a technological approach that can be used to uncover the mechanisms of defective cellular pathways in many other contexts, the authors said.
"If there is ever going to be a drug that targets this pathway with the goal of treating Parkinson's or other neurodegenerative diseases, we are going to need this level of detail to understand how drug candidates work," said senior study author J. Wade Harper, the HMS Bert and Natalie Vallee Professor of Molecular Pathology and Chair of the Department of Cell Biology.
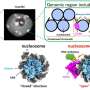
The molecular alarm system for mitochondrial quality control involves two enzymes: the protein kinase PINK1, which chemically modifies proteins with phosphate, and the ubiquitin ligase PARKIN, which marks target proteins with a molecule called ubiquitin.
Under normal conditions, healthy mitochondria carry little PINK1 on their outer surface. When damaged, however, mitochondria accumulate PINK1, which transfers phosphate to PARKIN to activate it.
Once activated, PARKIN transfers ubiquitin to a variety of different proteins, creating a "ubiquitin coat" on the mitochondrial surface. When a certain threshold of ubiquitin is reached, the recruitment of the cellular "self-eating" machinery is triggered, leading to degradation of the mitochondria.
Mutations in both PINK1 and PARKIN have been linked by numerous studies to Parkinson's disease, which involves the death of certain neurons and a buildup of improperly folded proteins in the brain. Researchers have made great strides in describing the PINK1-PARKIN pathway, but many of its critical features have remained poorly understood.
Tailoring the coat
Ubiquitin is known to mark its target proteins on specific residues of the amino acid lysine. This occurs for only a small subset of lysine residues, however, and determining precisely which lysines are modified and why has been a challenge for the field—especially for the more than a dozen PARKIN targets on the mitochondrial surface. In addition, the abundance of these targets was unclear, which could impact the ubiquitylation process.
Previous work by the Harper lab, in collaboration with Steve Gygi, HMS professor of cell biology, had established methods that allow detection of individual lysine residues in proteins that are linked with ubiquitin.
The research team extended this approach in the current study. Spearheaded by lead author Alban Ordureau, HMS research fellow in cell biology, they developed a new method—PARKIN target-parallel reaction monitoring (Pt-PRM)—to assess the landscape of protein ubiquitylation involved in mitochondrial quality control at an unprecedented level of detail.
They also created a library of reference peptides that allowed them to measure the precise abundance of PARKIN targets and individual ubiquitylation events on damaged mitochondria.
In essence, the reference peptide library enabled the researchers to "count" ubiquitylation events in a site-specific manner across more than a dozen mitochondrial surface proteins simultaneously in a mass spectrometer.
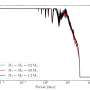
When combined with time-dependent measurements after mitochondrial damage, the method produces a "digital snapshot" of ubiquitylation events across the mitochondrial surface, revealing not only the site specificity of modification but also providing a quantitative picture of the mitochondrial landscape.
By including reference peptides that detect PINK1-dependent phosphorylation events, the team was also able to integrate target phosphorylation with ubiquitylation.
"This is one of the most detailed analyses of the ubiquitin pathway that's ever been done. There's no other study that uses this level of sophistication to analyze such a process," Harper said.
Digital snapshot
The new technique allows for accurate, absolute measurements of protein ubiquitylation events, in stark contrast to previous methods that are nonquantitative and do not provide ubiquitylation-site specificity.
"This approach now gives us the full dynamic range," Harper said. "We can measure over three orders of magnitude to better understand kinetics, stoichiometry, spatial organization, integration with phosphorylation and many other features," Harper said.
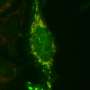
Enabled by improved precision, the team performed the first quantitative analysis of native ubiquitylation-related events in neurons derived from genetically modified human stem cells, a system more relevant to the study of human disease than previous models.
This included an analysis of dopaminergic neurons, the loss of which is a primary characteristic of Parkinson's, done in collaboration with Lee Rubin, professor of stem cell and regenerative biology at Harvard. Thus far, most research in this area has been conducted on immortalized human cancer cells (HeLa) engineered to express PARKIN at unnaturally high levels so that scientists could measure their activity.
Harper, Ordureau and colleagues compared the two cell models in their analyses, identifying similarities and differences in the specificity and timing of modification events. They also found specific lysine residues on proteins that were selectively ubiquitylated in neurons but not in HeLa cells, and vice versa. The biological implications of these differences remain unknown, and are targets for further study, the authors said.
The analyses helped shed new light on currently debated questions in the field, such as whether ubiquitin chains indiscriminately tag proteins on the mitochondria or whether they precision-target specific ones.
The answer may be "it depends." In general, ubiquitylation appears to scale with the abundance of protein, but the researchers found evidence for selectivity as well, particularly within certain structural elements within the target proteins.
The robustness of the experimental system also allowed the authors to examine several mechanistic aspects of the pathway, including, for example, a determination of the role of PARKIN phosphorylation by PINK1 in stem-cell derived neurons.
"We want to know how these mechanisms work because we want to be able to target them for drug discovery, or find ways to bypass disease-causing mutations in patients," Ordureau said. "We can now analyze this critical process in neuronal cells in extreme detail, and do so at scale in one experiment. With previous methods, you had to run dozens of experiments to achieve similar or less accurate results."
The efficacy of their new method now enables improved studies of the PINK1-PARKIN pathway and of its role in diseases such as Parkinson's, as demonstrated in this work for small-molecule activators of PINK1. The approach can also be adapted to uncover the mechanisms of defective pathways in other systems and diseases, the authors said.
"Ubiquitylation is a foundational mechanism that cells use to remove damaged or unneeded components to maintain their own health and integrity," Harper said. "It's relevant not only for mitochondria, but for removal of bacteria, protein aggregates and other organelles. This technology could be employed to shed light on the biochemistry of many different systems."
More information: Alban Ordureau et al, Dynamics of PARKIN-Dependent Mitochondrial Ubiquitylation in Induced Neurons and Model Systems Revealed by Digital Snapshot Proteomics, Molecular Cell (2018). DOI: 10.1016/j.molcel.2018.03.012
Journal information: Molecular Cell
Provided by Harvard Medical School