Mitochondria mutation mystery solved: Random sorting helps get rid of duds

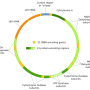
You probably know about the 23 pairs of chromosomes safely stowed in your cells' nuclei. That's where the vast majority of your genes can be found. But there are 37 special genes—a very tiny fraction of the human genome—located in mitochondria, the structures inside your cells that breathe and produce energy.
Repeated copying of mitochondrial DNA introduces errors; if not kept in check, these mutations can give rise to incurable diseases like Leigh syndrome and Leber's optic neuropathy. Worldwide, more than 1 in 10,000 people are affected by disorders resulting from mitochondrial genome defects.
Unlike nuclear chromosomes that we get from both parents, only mothers' mitochondria are passed on to offspring. This makes the usual process of sexual recombination, in which pieces of maternal and paternal chromosomes combine to repair genome defects, impossible. For decades, biologists predicted that without this repair mechanism, mitochondrial genes should rapidly accumulate harmful mutations and lose their function.
Despite these predictions, mitochondrial disorders in humans, while debilitating, are relatively rare. A set of experiments with human embryos has recently found low levels of mitochondrial mutations in most of the studied cells, that, strikingly, were otherwise perfectly healthy. If mitochondrial defects are so common, what keeps them from reaching dangerous disease-causing levels?

Dealing out mitochondria by chance
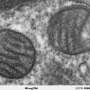
A typical human cell contains hundreds of mitochondria. Each mitochondrion in turn has many genome copies jointly responsible for energy production. If only a few of these copies become faulty, the rest of the mitochondria can still produce enough energy, and the cell does perfectly fine. In fact, some of the most severe disorders develop only when 60 to 90 percent of mitochondria within each cell become mutated. This means that low levels of mitochondrial mutations are essentially invisible, and can lurk within human cells for generations without causing a disease.
Recent theoretical work by my colleagues and me predicted a number of solutions that likely evolved to expose and eventually eliminate these hidden defects. The general principle we proposed is based on simple sorting of healthy and faulty mitochondria.
Whenever a cell within a developing embryo divides, mitochondria are partitioned into the two daughter cells more or less randomly. By chance, one of the two daughter cells inherits more mitochondrial defects than the other. Initially, this difference is barely noticeable. But repeat the process many times and a sizeable proportion of all daughter cells will have enough mutations to ensure that the cell does not survive.
On the opposite side of the spectrum, this leaves cells that have fewer mutations even than the original cell that started dividing. This simple mechanism of cell division and random sorting of mitochondria can therefore produce cells packed with healthy mitochondria that can then go on to divide further and to eventually produce mutation-free reproductive cells (eggs in females).

But there's more. Scientists now believe that many features of the human reproductive system evolved to increase the efficiency of this random mitochondrial sorting. For instance, mutations would pile up faster if both paternal and maternal mitochondria were inherited by the offspring – mixing of two unrelated types of organelles would make it easier for rare defects to hide. It is very likely that we inherit mitochondrial genes only from our mothers precisely because it slows down the accumulation of defective genes.
The number of genome replication cycles also matters, because new defects are introduced each time genes are copied. In a paper published in 2016, my colleagues and I suggest this could be the reason why the number of cell divisions to produce an egg in females is strictly limited to 24. In males – whose mitochondria are not transmitted to the offspring – sperm are produced continually with more than 400 cell divisions by the age of 30. By capping the number of times a cell divides before an egg is made, females reduce the risk of introducing new copying errors in their mitochondrial genes.
Likewise, theory predicts that random sorting of healthy and sickly mitochondria works best when the number of mitochondria in a cell is low. With only a few mitochondria, even slightly defective genes cannot hide; their harmful effects are immediately obvious at the level of the cell, which can then be eliminated.
Observing what theory predicts
Confirming these predictions, a recent study involving human embryos has indeed discovered that the number of mitochondria is sharply reduced throughout development – from 1 million in a fertilized egg to only around 1,500 per cell in a 4-week-old embryo. Researchers also found that cells taken from older embryos had fewer mitochondrial mutations, meaning that cells with the most defects were somehow eliminated throughout embryonic development.
It is not yet clear how cells with the most mitochondrial mutations are selectively removed in human embryos. But because most of the harmful mutations were eliminated at the stage of embryonic development when cells start breathing more actively, scientists think that damaged mitochondria simply fail to produce enough energy for the cell to survive.
Many questions remain. For instance, why do cells with high levels of defective mitochondria sometimes escape these quality-control mechanisms, resulting in incurable disorders? Ultimately, greater understanding of these mechanisms should suggest better ways of estimating the risk of mitochondrial diseases, or even develop new interventions to prevent them completely.
Provided by The Conversation
This article was originally published on The Conversation. Read the original article.![]()