Few-layer tellurium as a promising successor of black phosphorus
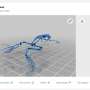
 vibrational behaviors, higher mobility along the non-covalent bonding direction and isotropic light absorption, as governed by the found covalent-like quasi-bonding for the inter-chain (-layer) interactions. Credit: Science China Press")
Two-dimensional (2-D) layered materials have received considerable attention for their potential applications since the experimental discovery of graphene. Theoretical two-dimensional elementary semiconductors promise superior features in terms of fabrication, purification and doping. Few-layer black phosphorus (BP) is the first 2-D mono-elementary semiconductor with high electronic carrier mobility, strong optical absorption, linear dichroism, and high tunability with external fields. However, the flawed air stability and difficulties in large scale fabrication are remaining issues that inhibit practical applications of few-layer BP. Thus, researchers seek possible alternatives that also allow low-cost, large-scale synthesis, and offer good environmental stability without sacrificing the advantages of BP.
Professor Wei Ji and his research group at Renmin University of China theoretically modeled surfaces and interfaces of emerging electronic materials to predict physical properties of devices composed of these materials. Recently, they collaborated with Prof. Yang Chai from the Hong Kong Polytechnic University to report a theoretical study of a novel, chain-like 2-D-material, namely few-layer α-tellurium (FL-α-Te), and predicted this material to have extremely high carrier mobility with a layer-tunable bandgap, strong light absorption, mixing of vibrational modes, layer-dependent energy maps of valence and conduction bands, among other striking properties.
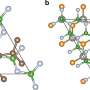
The FL-α-Te is a representative material of layered one-dimensional materials, which are a novel and fast-developing category of 2-D-materials. They first examined the stability of three likely few-layer phases using state-of-the-art density functional theory calculations. Their calculation shows α-tellurium is the most stable phase for bilayer and thicker layers. Given this stability, they found that a covalent-like quasi-bonding (CLQB) dominates the inter-chain interaction in both intra- and inter-layer directions. This CLQB is analogous to the found interlayer interactions in BP, PtS2 or PtSe2, showing wavefunction hybridization but without extra energy gain.
They managed to correlate this bonding with the layer-dependent geometric and electronic structures and their resulting behaviors in terms of electric, optical and vibrational properties. Few-layer α-Te has extremely high hole mobility up 105 cm2/Vs exceptionally along the non-covalent-bound (CLQB) direction and 103 cm2/Vs for the covalent-bound direction, tunable bandgap from 0.31 eV (Bulk) to 1.17 eV (2L), anisotropic inter-chain (-layer) vibrational behaviors, a crossover of interlayer shear and breathing force constants, large ideal strength (over 20 percent) and nearly isotropic strong light absorption (up to 9 percent per layer) from a highly anisotropic geometry. They also found within few-layer α-Te that the energy surfaces of both valence and conduction bands substantially develop from bulk to bilayer, exhibiting an "M-like" line profile of the hole pocket, which was usually found in topological insulators, and is ideal for thermoelectrics.
This material offers most of the striking properties of BP along with better environmental stability, a much lower fabrication cost (with wet-chemistry methods) and higher light absorption than those of BP. In this scenario, FL-α-Te could be regarded as a superior successor of BP. The extraordinarily high carrier mobility revealed in the CLQB direction conceptually updates the understanding of the role of non-covalent interactions in carrier mobility and may open a new avenue for seeking high carrier mobility materials.
More information: Jingsi Qiao et al, Few-layer Tellurium: one-dimensional-like layered elementary semiconductor with striking physical properties, Science Bulletin (2018). DOI: 10.1016/j.scib.2018.01.010
Provided by Science China Press