How robot math and smartphones led researchers to a drug discovery breakthrough

For us humans, a healthy brain handles all the minute details of bodily motion without demanding conscious attention. Not so for brainless robots – in fact, calculating robotic movement is its own scientific subfield.
My colleagues here at the University of Washington's Institute for Protein Design have figured out how to apply an algorithm originally designed to help robots move to an entirely different problem: drug discovery. The algorithm has helped unlock a class of molecules known as peptide macrocycles, which have appealing pharmaceutical properties.
One small step, one giant leap
Roboticists who program movement conceive of it in what they call "degrees of freedom." Take a metal arm, for instance. The elbow, wrist and knuckles are movable and thus contain degrees of freedom. The forearm, upper arm and individual sections of each finger do not. If you want to program an android to reach out and grasp an object or take a calculated step, you need to know what its degrees of freedom are and how to manipulate them.
The more degrees of freedom a limb has, the more complex its potential motions. The math required to direct even simple robotic limbs is surprisingly abstruse; Ferdinand Freudenstein, a father of the field, once called the calculations underlying the movement of a limb with seven joints "the Mount Everest of kinematics."
Freudenstein developed his kinematics equations at the dawn of the computer era in the 1950s. Since then, roboticists have increasingly relied on algorithms to solve these complex kinematic puzzles. One algorithm in particular – known as "generalized kinematic closure" – bested the seven joint problem, allowing roboticists to program fine control into mechanical hands.
Molecular biologists took notice.
Many molecules inside living cells can be conceived of as chains with pivot points, or degrees of freedom, akin to tiny robotic arms. These molecules flex and twist according to the laws of chemistry. Peptides and their elongated cousins, proteins, often must adopt precise three-dimensional shapes in order to function. Accurately predicting the complex shapes of peptides and proteins allows scientists like me to understand how they work.
Mastering macrocycles
While most peptides form straight chains, a subset, known as macrocycles, form rings. This shape offers distinct pharmacological advantages. Ringed structures are less flexible than floppy chains, making macrocycles extremely stable. And because they lack free ends, some can resist rapid degradation in the body – an otherwise common fate for ingested peptides.
Natural macrocycles such as cyclosporin are among the most potent therapeutics identified to date. They combine the stability benefits of small-molecule drugs, like aspirin, and the specificity of large antibody therapeutics, like herceptin. Experts in the pharmaceutical industry regard this category of medicinal compounds as "attractive, albeit underappreciated."
"There is a huge diversity of macrocycles in nature – in bacteria, plants, some mammals," said Gaurav Bhardwaj, a lead author of the new report in Science, "and nature has evolved them for their own particular functions." Indeed, many natural macrocycles are toxins. Cyclosporin, for instance, displays anti-fungal activity yet also acts as a powerful immunosuppressant in the clinic making it useful as a treatment for rheumatoid arthritis or to prevent rejection of transplanted organs.
A popular strategy for producing new macrocycle drugs involves grafting medicinally useful features onto otherwise safe and stable natural macrocycle backbones. "When it works, it works really well, but there's a limited number of well-characterized structures that we can confidently use," said Bhardwaj. In other words, drug designers have only had access to a handful of starting points when making new macrocycle medications.
To create additional reliable starting points, his team used generalized kinematic closure – the robot joint algorithm – to explore the possible conformations, or shapes, that macrocycles can adopt.
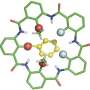
 and many ‘side chains’ (shown as thin lines). The macrocycle on the left — cyclosporin — evolved in a fungus. The one on the right was designed on a computer. Credit: Ian Haydon/Institute for Protein Design")
Adaptable algorithms
As with keys, the exact shape of a macrocycle matters. Build one with the right conformation and you may unlock a new cure.
Modeling realistic conformations is "one of the hardest parts" of macrocycle design, according to Vikram Mulligan, another lead author of the report. But thanks to the efficiency of the robotics-inspired algorithm, the team was able to achieve "near-exhaustive sampling" of plausible conformations at "relatively low computational cost."
The calculations were so efficient, in fact, that most of the work did not require a supercomputer, as is usually the case in the field of molecular engineering. Instead, thousands of smartphones belonging to volunteers were networked together to form a distributed computing grid, and the scientific calculations were doled out in manageable chunks.
With the initial smartphone number crunching complete, the team pored over the results – a collection of hundreds of never-before-seen macrocycles. When a dozen such compounds were chemically synthesized in the lab, nine were shown to actually adopt the predicted conformation. In other words, the smartphones were accurately rendering molecules that scientists can now optimize for their potential as targeted drugs.
The team estimates the number of macrocycles that can confidently be used as starting points for drug design has jumped from fewer than 10 to over 200, thanks to this work. Many of the newly designed macrocycles contain chemical features that have never been seen in biology.
To date, macrocyclic peptide drugs have shown promise in battling cancer, cardiovascular disease, inflammation and infection. Thanks to the mathematics of robotics, a few smartphones and some cross-disciplinary thinking, patients may soon see even more benefits from this promising class of molecules.
Journal information: Science
Provided by The Conversation
This article was originally published on The Conversation. Read the original article.![]()