By modeling biological molecules over longer timescales, a new algorithm can help better understand diseases
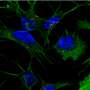
. The fibrillar aggregates act as cell-toxins at the onset and the progression of Alzheimer's disease. Credit: Emanuel Peter")
Proteins, the ubiquitous workhorses of biochemistry, are huge molecules whose function depends on how they fold into intricate structures. To understand how these molecules work, researchers use computer modeling to calculate how proteins fold.
Now, a new algorithm can accelerate those vital simulations, enabling them to model phenomena that were previously out of reach. The results can eventually help scientists better understand and treat diseases like Alzheimer's, said Emanuel Peter, a chemist at the University of Regensburg. His work on the new technique is described this week in The Journal of Chemical Physics.
Conventional simulations, using molecular dynamics and Monte Carlo methods, have been successful overall at modeling biological molecules like proteins. To determine how proteins fold, the simulation searches for configurations that correspond to lower and lower energy states. Eventually, it finds the lowest energy state, which gives a stable fold. But as the simulation searches, it may encounter a configuration with a slightly higher energy, which forms a barrier that impedes the algorithm.
As a result of these slowdowns, conventional methods can only simulate molecular behaviors occurring over short time scales of a few hundred microseconds. Many phenomena, such as certain protein folds or a drug binding to a potential target, happen over the course of a few seconds, minutes or even days. Simulating such long timescales would take too much computation time with just conventional approaches.
To speed up the simulations, researchers can inject energy into the system, which pushes the model over any energy barriers. But one of the biggest challenges to these methods is in defining the coordinates that describe the system—which, for example, can be the length between atoms in the molecule, and the angles between bonds. Traditionally, researchers define the coordinates before they start the simulation. Every time step along each coordinate depends on the previous step. But this dependency can bias the simulation.
Peter's new algorithm avoids this bias. He found a generalized coordinate system in which each time step doesn't rely on the previous step. "Only few parameters are needed, and no human intuition is required, which can potentially bias the simulation result," he said.
To test the new algorithm, Peter used it to model water, a peptide called dialanine, the folding of another peptide called TrpCage, and the clumping of amyloid-beta 25-35, which are protein fragments associated with Alzheimer's disease. In each case, his technique reports to have sped up the simulations. And the simulations of amyloid-beta could help explain why Alzheimer's has been difficult to treat.
In Alzheimer's disease, amyloid-beta protein fragments aggregate together, forming hard plaque that builds up between neurons and disrupts them. Amyloid-beta is also a toxin, leading to neuronal cell death and degeneration of neuronal function. The new simulations suggest that amyloid-beta can assume a range of structures. This structural flexibility could be why some drugs that try to inhibit aggregation haven't been successful, Peter said. When those drugs bind to amyloid-beta, the amyloid-beta just changes shape, allowing it to continue clumping together. The drug becomes incorporated into the aggregate and the plaque.
This type of structural flexibility, called conformation entropy, is also a key feature in other peptides that form toxic plaques in diseases such as Huntington's disease, Type 2 diabetes, and Parkinson's disease. The new algorithm could therefore be useful for understanding these other diseases as well.
More information: "Adaptive enhanced sampling with a path-variable for the simulation of protein folding and aggregation," Journal of Chemical Physics (2017). DOI: 10.1063/1.5000930
Journal information: Journal of Chemical Physics
Provided by American Institute of Physics