Team reconstructs nanoscale virus features from correlations of scattered X-rays
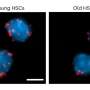
 Peter Zwart, Jeff Donatelli and Kanupriya Pande, co- authors of a paper describing how the group's M-TIP algorithm determined 3D virus structures from single-particle diffraction data. Donatelli holds a 3D-printed model of one of the viruses reconstructed by M-TIP. Credit: Marilyn Chung, Berkeley Lab")
As part of an international research team, Jeff Donatelli, Peter Zwart and Kanupriya Pande of the Center for Advanced Mathematics for Energy Research Applications (CAMERA) at Lawrence Berkeley National Laboratory (Berkeley Lab) contributed key algorithms which helped achieve a goal first proposed more than 40 years ago - using angular correlations of X-ray snapshots from non-crystalline molecules to determine the 3D structure of important biological objects. This technique has the potential to allow scientists to shed light on biological structure and dynamics that were previously impossible to observe with traditional X-ray methods.
The breakthrough resulted from a single-particle diffraction experiment conducted at the Department of Energy's (DOE's) Linac Coherent Light Source (LCLS) by the Single-Particle Initiative organized by the SLAC National Accelerator Laboratory. As part of this initiative, the CAMERA team combined efforts with Ruslan Kurta, a physicist at the European XFEL (X-ray free electron laser) facility in Germany, to analyze angular correlations from the experimental data and use CAMERA's multi-tiered iterative phasing (M-TIP) algorithm to perform the first successful 3D virus reconstructions from experimental correlations. The results were described in a paper published Oct. 12 in Physical Review Letters.
"For the past 40 years, this was considered a problem that could not be solved," said Peter Zwart, co-author on the paper and a physical bioscientist who is a member of CAMERA based out of the Molecular Biophysics and Integrated Imaging Division at Berkeley Lab. "But it turns out that the mathematical tools that we developed are able to leverage extra information hidden in the problem that had been previously overlooked. It is gratifying to see our theoretical approach lead to a practical tool."
New Research Opportunities Enabled by XFELs
For much of the last century, the go-to technique for determining high-resolution molecular structure has been X-ray crystallography, where the sample of interest is arranged into a large periodic lattice and exposed to X-rays which scatter off and form diffraction patterns that are collected on a detector. Even though crystallography has been successful at determining many high-resolution structures, it is challenging to use this technique to study structures which are not susceptible to crystallization or structural changes that do not naturally occur within a crystal.
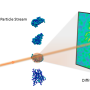
The creation of XFEL facilities, including the Linac Coherent Light Source (LCLS) and the European X-FEL, have created opportunities for conducting new experiments which can overcome the limitations of traditional crystallography. In particular, XFEL beams are several orders of magnitude brighter than and have much shorter pulse lengths than traditional X-ray light sources, which allow them to collect measurable diffraction signal from smaller uncrystallized samples and also study fast dynamics. Single-particle diffraction is one such emerging experimental technique enabled by XFELS, where one collects diffraction images from single molecules instead of crystals. These single-particle techniques can be used to study molecular structure and dynamics that have been difficult to study with traditional imaging techniques.
Overcoming Limitations in Single-Particle Imaging via Angular Correlations
One major challenge of single-particle imaging is that of orientation determination. "In a single-particle experiment, you don't have control over rotation of the particles as they are hit by the X-ray beam, so each snapshot from a successful hit will contain information about the sample from a different orientation," said co-author Jeff Donatelli, an applied mathematician in CAMERA who developed many of the algorithms in the new framework. "Most approaches to single-particle analysis have so far been based on trying to determine these particle orientations from the images; however, the best resolution achievable from these analyses is restricted by how precisely these orientations can be determined from noisy data."
Instead of trying to directly determine these orientations, the team took a different approach based on idea originally proposed in the 1970s by Zvi Kam. "Rather than examine the individual data intensities in an attempt to find the correct orientation for each measured frame, we eliminate this step altogether by using so-called cross-correlation functions," Kurta said.
This approach, known as fluctuation X-ray scattering, is based on analyzing the angular correlations of ultrashort, intense X-ray pulses scattered from non-crystalline biomolecules. "The beauty of using correlation data is that it contains a comprehensive fingerprint of the 3D structure of an object that extends traditional solution scattering approaches," Zwart said.
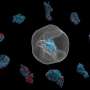
 and a PR772 bacteriophage (bottom) from experimental correlation data using M-TIP. The images on the right show asymmetries in the internal genetic material for each virus reconstruction. Credit: Jeff Donatelli, Berkeley Lab")
Reconstructing 3D Structure from Correlations with CAMERA's M-TIP Algorithm
The team's breakthrough in reconstructing 3D structure from correlation data was enabled by the multi-tiered iterative phasing (M-TIP) algorithm developed by CAMERA. "Among the prominent advantages of M-TIP is its ability to solve the structure directly from the correlation data without having to rely on any symmetry constraints, and, more importantly, without the need to solve the orientation determination problem," Donatelli said.
Donatelli, CAMERA director James Sethian and Zwart developed their M-TIP framework by developing a mathematical generalization of a class of algorithms known as iterative phasing techniques, which are used for determining structure in a simpler problem, known as phase retrieval. A paper describing the original M-TIP framework was published August 2015 in the Proceedings of the National Academy of Sciences.
"Advanced correlation analyses in combination with ab-initio reconstructions by M-TIP clearly define an efficient route for structural analysis of nanoscale objects at XFELs," Zwart said.

Future Directions for Correlation Analysis and M-TIP
The team notes that methods used in this analysis can also be applied to analyze diffraction data when there is more than one particle per shot.
"Most algorithms for single-particle imaging can only handle one molecule at a time, thus limiting signal and resolution. Our approach, on the other hand, is scalable so that we should also be able to measure more than one particle at a time," said Kurta. Imaging with more than one particle per shot will allow scientists to achieve much higher hit rates, since it is easier to use a wide beam and hit many particles at a time, and will also avoid the need to separate out single-particle hits from multiple-particle hits and blank shots, which is another challenging requirement in traditional single-particle imaging.
As part of CAMERA's suite of computational tools, they have also developed a different version of M-TIP which solves the orientation problem and can classify the images into conformational states, and consequently can used to study small biological differences in the measured sample. This alternate version of M-TIP was described in a paper published June 26 2017 in the Proceedings of the National Academy of Sciences and is part of a new collaboration initiative between SLAC National Accelerator Laboratory, CAMERA, the National Energy Research Scientific Computing Center (NERSC) and Los Alamos National Laboratory as part of DOE's Exascale Computing Project (ECP).
More information: Ruslan P. Kurta et al. Correlations in Scattered X-Ray Laser Pulses Reveal Nanoscale Structural Features of Viruses, Physical Review Letters (2017). DOI: 10.1103/PhysRevLett.119.158102
Journal information: Physical Review Letters , Proceedings of the National Academy of Sciences
Provided by Lawrence Berkeley National Laboratory