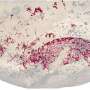
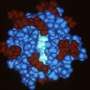
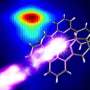
Models from big molecules captured in a flash
 using existing diffraction data superposed on known high-resolution structures. Top left, lysine-arginine-ornithine (LAO) binding protein; top right, lysozome; bottom left, peroxiredoxin; and, bottom right, Satellite Tobacco Mosaic Virus (STMV).")
(Phys.org) —To learn how biological molecules like proteins function, scientists must first understand their structures. Almost as important is understanding how the structures change, as molecules in the native state do their jobs.
Existing methods for solving structure largely depend on crystallized molecules, and the shapes of more than 80,000 proteins in a static state have been solved this way. The majority of the two million proteins in the human body can't be crystallized, however. For most of them, even their low-resolution structures are still unknown.
Their chance to shine may have come at last, thanks to new techniques developed by Peter Zwart and his colleagues at the U.S. Department of Energy's Lawrence Berkeley National Laboratory (Berkeley Lab), working with collaborators from Arizona State University, the University of Wisconsin-Milwaukee, and DOE's Pacific Northwest National Laboratory (PNNL). The new method promises a more informative look at large biological molecules in their native, more fluid state.
The researchers describe their results in two recent papers in Foundations of Crystallography and in Physical Review Letters.
Diffraction before destruction
A key factor in new ways of looking at biomolecules is the data created by free-electron lasers (FELs) such as the Linac Coherent Light Source (LCLS) at SLAC National Accelerator Laboratory, whose powerful x‑ray pulses are measured in quadrillionths of a second. These pulses are faster than a molecule can rotate, and the experimental data reflects the state of the molecule frozen in time.

"It's a technique called 'diffract before destroy,' because the data is collected before the particle literally blows apart," says Zwart, a member of the Lab's Physical Biosciences Division, and the science lead for the Berkeley Center for Structural Biology at the Advanced Light Source. "FELs have shown they can derive structures from single particles, each hit with a single pulse, but there are major challenges to this approach."
Instead of single particles, Zwart and his colleagues include many particles in each shot. When analyzed by computer programs, the data from the different diffraction patterns can be combined to provide detailed insights into the structures the molecules adopt in solution.
The technique is called fluctuation x-ray scattering (fXS), and Zwart and his colleagues have shown that data obtained this way with free-electron lasers can yield low-resolution shapes of biomolecules in close to their natural state, with much greater confidence than is currently possible with less powerful synchrotron light sources.
"Our algorithm starts with a trial model and modifies it by randomly adding or subtracting volume until the shape of the model achieves the optimum fit with the data," Zwart says. This trial-and-error optimization technique, tested on known configurations at the LCLS, can resolve the shapes of individual macromolecules with fXS data alone.
It's not only the structures of molecules taken one at a time that can be solved this way. Zwart and his former postdoc Gang Chen, working with Dongsheng Li of PNNL, have shown that data from mixtures of different kinds of molecules can be untangled to provide clues on the structure of the individual components, forming a basis for understanding the dynamic behavior of large biological molecules working together in solution.
By understanding their structural changes, Zwart and his colleagues are developing fluctuation x-ray scattering as an indispensable tool for determining how mixtures of different proteins behave independently or in concert.
More information: "Three-dimensional single-particle imaging using angular correlations from X-ray laser data," by Haiguang Liu, Billy K. Poon, Dilano K. Saldin, John C. H. Spence, and Peter H. Zwart, appears in the July 2013 issue of Foundations of Crystallography and is available online at journals.iucr.org/a/issues/201 … /04/00/issconts.html
"Component particle structure in heterogeneous disordered ensembles extracted from high-throughput fluctuation x-ray scattering," by Gang Chen, Peter H. Zwart, and Dongsheng Li, appears in the 10 May 2013 edition of Physical Review Letters, and is available online at prl.aps.org/abstract/PRL/v110/i19/e195501
Journal information: Physical Review Letters
Provided by Lawrence Berkeley National Laboratory