A fresh math perspective opens new possibilities for computational chemistry

Glow-in-the-dark objects seem magical when you're a kid—they can brighten up a dark room without the need for electricity, batteries or a light bulb. Then at some point you learn the science behind this phenomenon. Chemical compounds called chromophores become energized, or excited, when they absorb visible light. As they return to their normal state, the stored energy is released as light, which we perceive as a glow. In materials science, researchers rely on a similar phenomenon to study the structures of materials that will eventually be used in chemical catalysis, batteries, solar applications and more.
When a molecule absorbs a photon—the fundamental particle of light—electrons in the molecular system are promoted from a low-energy (ground) state to a higher-energy (excited) state. These responses resonate at specific light frequencies, leaving "spectral fingerprints" that illuminate the atomic and electronic structures of the system being studied.
In experiments, the "spectral fingerprints" or absorption spectrum, are measured with state-of-the-art facilities like the Advanced Light Source (ALS) at the U.S. Department of Energy's Lawrence Berkeley National Laboratory (Berkeley Lab). In computer simulations, these measurements are typically captured with a quantum mechanical method called Time Dependent Density Functional Theory (TDDFT). The computational models are critical in helping researchers make the most of their experiments by predicting and validating results.
Yet despite its usefulness, there are times when TDDFT cannot not be used to calculate the absorption spectrum of a system because it would require too much time and computer resources. This is where a new mathematical "shortcut" developed by researchers in Berkeley Lab's Computational Research Division (CRD) comes in handy. Their algorithm speeds up absorption calculations by a factor of five, so simulations that used to take 10 to 15 hours to compute can now be done in approximately 2.5 hours.
A paper describing this method was published in the Journal of Chemical Theory and Computation (JCTC). And the new approach for computing the absorption spectrum will be incorporated in an upcoming release of the widely used NWChem computational chemistry software suite later this year.
New Algorithms Lead to Computational Savings
To study the chemical structure of new molecules and materials, scientists typically probe the system with an external stimulus—typically a laser—then look for small electronic changes. Mathematically, this electronic change can be expressed as an eigenvalue problem. By solving this eigenvalue problem, researchers can get a good approximation of the absorption spectrum, which in turn reveals the resonant frequencies of the system being studied. Meanwhile, the corresponding eigenvector is used to calculate how intensely the system responded to the stimulus. This is essentially the principle behind the TDDFT approach, which has been implemented in several quantum chemistry software packages, including the open-source NWChem software suite.
While this approach has proven to be successful, it does have limitations for large systems. The wider the energy range of electronic responses a researcher tries to capture in a system, the more eigenvalues and eigenvectors need to be computed, which also means more computing resources are necessary. Ultimately, the absorption spectrum of a molecular system with more than 100 atoms becomes prohibitively expensive to compute with this method.

To overcome these limitations, mathematicians in CRD developed a technique to compute the absorption spectrum directly without explicitly computing the eigenvalues of the matrix.
"Traditionally, researchers have had to compute the eigenvalues and eigenvectors of very large matrices in order to generate the absorption spectrum, but we realized that you don't have to compute every single eigenvalue to get an accurate view of the absorption spectrum," says Chao Yang, a CRD mathematician who led the development of the new approach.
By reformulating the problem as a matrix function approximation, making use of a special transformation and taking advantage of the underlying symmetry with respect to a non-Euclidean metric, Yang and his colleagues were able to apply the Lanczos algorithm and a Kernal Polynomial Method (KPM) to approximate the absorption spectrum of several molecules. Both of these algorithms require relatively low-memory compared to non-symmetrical alternatives, which is the key to the computational savings.
Because this method requires less computing power to achieve a result, researchers can also easily calculate the absorption spectrum for molecular systems with several hundreds of atoms.
"This method is a significant step forward because it allows us to model the absorption spectrum of molecular systems of hundreds of atoms at lower computational cost." says Niranjan Govind, a computational chemist at the Pacific Northwest National Laboratory who collaborated with the Berkeley Lab team on the development of the method in the NWChem computational chemistry program.
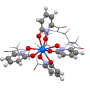
Recently Berkeley Lab scientists used this method to calculate the absorption spectrum and confirm what several experimental results have been hinting—that the element berkelium breaks form with its heavy element peers by taking on an extra positive charge when bound to a synthetic organic molecule. This property could help scientists develop better methods for handling and purifying nuclear materials. A paper highlighting this result appeared April 10 in the journal Nature Chemistry.
"The experimental results were hinting at this unusual behavior in berkelium, but there wasn't enough experimental evidence to say yes, 100 percent, this is what we're seeing," says study co-author Wibe Albert de Jong, a CRD scientist. "To be 100 percent sure, we did large computational simulations and compared them to the experimental data and determined that they were, indeed, seeing berkelium in an unusual oxidation state."
This new algorithm was developed through a DOE Office of Science-supported Scientific Discovery through Advanced Computing (SciDAC) project focused on advancing software and algorithms for photochemical reactions. SciDAC projects typically bring together an interdisciplinary team of researchers to develop new and novel computational methods for tackling some of the most challenging scientific problems.
"The interdisciplinary nature of SciDAC is a very effective way to facilitate breakthrough science, as each team member brings a different perspective to problem solving," says Yang. "In this dynamical environment, mathematicians, like me, team up with domain scientists to identify computational bottlenecks, then we use cutting-edge mathematical techniques to address and overcome those challenges."
More information: Jiri Brabec et al. Efficient Algorithms for Estimating the Absorption Spectrum within Linear Response TDDFT, Journal of Chemical Theory and Computation (2015). DOI: 10.1021/acs.jctc.5b00887
Journal information: Nature Chemistry
Provided by Lawrence Berkeley National Laboratory