Computational method makes gene expression analyses more accurate
. The chromosome is X-shaped because it is dividing. Introns are regions often found in eukaryote genes that are removed in the splicing process (after the DNA is transcribed into RNA): Only the exons encode the protein. The diagram labels a region of only 55 or so bases as a gene. In reality, most genes are hundreds of times longer. Credit: Thomas Splettstoesser/Wikipedia/CC BY-SA 4.0")
A new computational method can improve the accuracy of gene expression analyses, which are increasingly used to diagnose and monitor cancers and are a major tool for basic biological research.
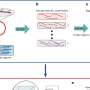
Researchers from Carnegie Mellon University, Stony Brook University and Dana-Farber Cancer Institute said their method, called Salmon, is able to correct for the technical biases that are known to occur during RNA sequencing, or RNA-seq, the leading method for estimating gene expression. Furthermore, it operates at similar speeds as other fast methods - a critical factor as these tests are growing more common and numerous.
Their report is being published online March 6 by the journal Nature Methods. Carl Kingsford, associate professor in CMU's Computational Biology Department, said the Salmon source code is freely available online and already has been downloaded by thousands of users.
"Salmon provides a much richer model of the RNA-seq experiment and of the possible biases that are known to occur during sequencing," Kingsford said. This is important, he added, because this technique is increasingly used for classifying diseases and their subtypes, understanding gene expression changes during development and tracking the progression of cancer.
Though an organism's genetic makeup is static, the activity of individual genes varies greatly over time, making gene expression an important factor in understanding how organisms work and what occurs during disease processes. Gene activity can't be efficiently measured directly, but can be inferred by monitoring RNA, the molecules that carry information from the genes for producing proteins and other cellular activities.
RNA-seq is a leading technology for producing these snapshots of gene activity. But depending on the tissue being analyzed and the way each sample is prepared, various experimental biases can occur and cause RNA-seq "reads" to be over- or under-sampled from various genes, Kingsford said.
"Though we know many of the kinds of biases that can occur, modeling them has to occur on a sample-by-sample basis," he added. "And if you have to build a complicated bias model using traditional methods, it takes a really long time."
The researchers named the method after a fish famous for swimming upstream because it employs an algorithm that can estimate the effect of biases and the expression level of genes as experimental data streams by.
"In that way, it is able to build up a rich bias model and do so approximately as fast as other fast analysis tools," Kingsford said.
More information: Salmon provides fast and bias-aware quantification of transcript expression, Nature Methods (2017). DOI: 10.1038/nmeth.4197
Journal information: Nature Methods
Provided by Carnegie Mellon University