Search technique helps researchers find DNA sequences in minutes rather than days
Database searches for DNA sequences that can take biologists and medical researchers days can now be completed in a matter of minutes, thanks to a new search method developed by computer scientists at Carnegie Mellon University.
The method developed by Carl Kingsford, associate professor of computational biology, and Brad Solomon, a Ph.D. student in the Computational Biology Department, is designed for searching so-called "short reads" - DNA and RNA sequences generated by high-throughput sequencing techniques. It relies on a new indexing data structure, called Sequence Bloom Trees, or SBTs, that the researchers describe in a report published online today by the journal Nature Biotechnology.
The National Institutes of Health maintains a humongous database, called the Sequence Read Archive, which contains about three petabases, or sequences totaling three quadrillion base-pairs. The information is useful to a wide swath of researchers, from those asking questions about basic biological processes to those studying potential cancer cures.
"The database contains untold numbers of as-yet undiscovered insights and is heavily used," Kingsford said. "Its main problem is that it's very difficult to search."
Thousands of hard drives would be needed to store these sequences. Searching through the short reads, which are typically 50 to 200 base-pairs each, to see which ones could be assembled to form a target gene of perhaps 10,000 base-pairs, is cumbersome and can take days in some cases, he noted.
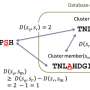
Just as an index can speed searches through a book or catalog, the SBT-based index developed by Kingsford and Solomon can greatly speedup searches of this bioinformatics database. They actually represent each short read as a set of fixed-length subsequences, employing data structures called Bloom filters that can efficiently store information in a small space and can test whether an element is part of a set.
At the first level of inquiry, the SBTs can tell whether a target DNA sequence is contained in the database at all. If it is, the search proceeds to the next level, where the SBTs indicate whether the sequence is in one half or the other of the database. At each level, the inquiry branches one way or the other until the desired experiments are identified.
Kingsford and Solomon tested their technique using a database of 2,652 human blood, breast and brain experiments, each of which often contain over a billion base-pairs of RNA sequences. They found that most searches of that database could be completed in an average of 20 minutes. They estimated the comparable search time using existing techniques, known as SRA-BLAST and STAR, would take 2.2 days and 921 days, respectively.
Further speedups are possible because batches of over 200,000 queries can be performed simultaneously, they noted.
More information: Fast search of thousands of short-read sequencing experiments, Nature Biotechnology, DOI: 10.1038/nbt.3442
The SBT method is available as open source code and can be downloaded at www.cs.cmu.edu/~ckingsf/software/bloomtree/
Journal information: Nature Biotechnology
Provided by Carnegie Mellon University