Seeking structure with metagenome sequences
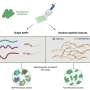
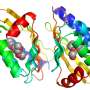
. Each position (or column) is compared to all other positions to detect patterns of co-evolution. Bottom: the strength of the top co-evolving residues is shown as blue dots, these are also shown as colored lines on the structure above. The goal is to make a structure that makes as many of these contacts as possible. Right: a cartoon of the protein structure predicted. The protein domain shown is from Pfam DUF3794, this domain is part of a Spore coat assembly protein SafA. (Image of Great Boiling Spring by Brian Hedlund, UNLV. Protein structure and composite image by Sergey Ovchinnikov, UW)")
For proteins, appearance matters. These important molecules largely form a cell's structures and carry out its functions: proteins control growth and influence mobility, serve as catalysts, and transport or store other molecules. Comprised of long amino acid chains, the one-dimensional amino acid sequence may seem meaningless on paper. Yet when viewed in three dimensions, researchers can see what a protein's structure is and how a protein's structure, and particularly the way it folds, determines its functions.
There are close to 15,000 protein families - groups of families that share an evolutionary origin - in the database Pfam. For nearly a third (4,752) of these protein families, there is at least one protein in each family that already has an experimentally determined structure. For another third (4,886) of the protein families, comparative models could be built with some degree of confidence. For the final third (5,211) of the protein families in the database, however, no structural information exists.
In the January 20, 2017 issue of Science, a team led by University of Washington's David Baker in collaboration with researchers at the U.S. Department of Energy Joint Genome Institute (DOE JGI), a DOE Office of Science User Facility, reports that structural models have been generated for 614 or 12 percent of the protein families that had previously had no structural information available. "That this could be accomplished using computational modeling methods was not at all apparent 5 years ago," the team noted in their paper. This accomplishment was made possible through a collaboration in which the Baker lab's protein structure prediction server Rosetta analyzed the metagenomic sequences publicly available on the Integrated Microbial Genomes (IMG) system run by the DOE JGI.
"A large number of protein families (in Pfam) have low number of sequences," said study first author Sergey Ovchinnikov, a graduate student in the Baker lab. "This resulted in two consequences: 1) nobody cared about these families (since they were small); and, 2) co-evolution methods could not be applied to study them. With metagenomics, we found that some of these neglected families with only a handful of sequences so far, can now become as large as some of the most studied ones, when metagenomics data are taken into account! Moreover, we can offer a 3D model of a representative sequence from the family. We hope this will spark interest in some of these families."
 there is always a complementary green amino acid on the right (and vise versa). This would indicate these two positions likely form a physical interaction, allowing us to draw the two structures on the right. Credit: Sergey Ovchinnikov, UW")
Armed with genome sequences, researchers like Baker have been able to identify sets of amino acids that evolve simultaneously, even though they are nowhere near each other on the unfolded chain. Such events suggests these amino acids are neighbors in the folded protein, offering researchers hints as to the protein's structure. Structural proximity can suggest a functional relationship and thus natural selection, acting on the function, can favor not just one amino acid but all that are in the set.
Nikos Kyrpides, DOE JGI Prokaryote Super Program head, said the collaboration between the Baker lab and the DOE JGI allowed the team to come up with a powerful way of predicting structures and structural alignments. "Such efforts, were previously restricted on protein families generated from sequences found on the isolate genome only. These genomes comprise about 200 million sequences. As expected, when we added on those our metagenomics data, harnessing the 5 billion assembled metagenome sequences available on our IMG/M database, we were able to dramatically increase the coverage of many of the known protein families. Efforts like this one heavily depend on the availability of assembled metagenomics sequences, which is an advantage the DOE JGI brings to the table with our high quality assemblies."
Kyrpides added that this work, which also involved DOE JGI researchers Neha Varghese and George Pavlopoulos, embodies another kind of collaboration that he'd like to see encouraged. "People came to us because we are maintaining the largest integration of assembled metagenomes. The application of such tools on our data provides a great example of how the larger community can utilize JGI resources for discovery. We would very much like to see more success stories like this one through a new Data Science call between the JGI and the National Energy Research Scientific Computing Center (NERSC)."
The JGI-NERSC Microbiome Data Science call will enable users to perform state-of-the-art computational genomics and metagenomics research and help them translate sequence information, generated by the DOE JGI or elsewhere, into biological discovery. This proposal call builds upon the success of "Facilities Integrating Collaborations for User Science" (FICUS) initiative, established to encourage and enable researchers to more easily integrate the expertise and capabilities of multiple national user facilities into their research. Applications for JGI-NERSC collaborative science call are currently being accepted until March 1, 2017. For more information about the call, go to: http://jgi.doe.gov/user-program-info/community-science-program/how-to-propose-a-csp-project/ficus-jgi-nersc/.
More information: Sergey Ovchinnikov et al, Protein structure determination using metagenome sequence data, Science (2017). DOI: 10.1126/science.aah4043
Journal information: Science
Provided by DOE/Joint Genome Institute