Improved method for protein sequence comparisons is faster, more accurate, sensitive
Lightning fast and yet highly sensitive: HHblits is a new software tool for protein research which promises to significantly improve the functional analysis of proteins. A team of computational biologists led by Dr. Johannes Soding of LMU's Genzentrum has developed a new sequence search method to identify proteins with similar sequences in databases that is faster and can discover twice as many evolutionarily related proteins as previous methods. From the functional and structural properties of the identified proteins conclusions can then be drawn on the properties of the protein to be analysed.
"Our method will expand the scope and power of sequence analysis, which will in turn facilitate the experimental elucidation of the structure and function of many proteins", says Söding, who is also a member of the Center for Integrated Protein Science Munich (CiPSM).
Proteins are involved in nearly all biochemical processes of life. The functions that a protein performs largely depend on the sequence of the 20 amino acid building blocks and on the three-dimensional spatial structure into which this sequence of amino acids folds. From the similarity of protein sequences, bioinformatic methods can predict their evolutionary relatedness, which in turn implies similar structure and functions. Therefore, proteins to be studied are standardly subjected to a sequence search, in which their sequence is compared with millions of sequences in public databases with annotated structures and functions. The properties of the protein of interest can then be inferred from the properties of the proteins with similar sequences, including its structure and functions. The general relationship between sequence and function makes it possible to predict the structure and function of a given protein by comparing its sequence with those of proteins of known structure/function. Publicly accessible databases exist in which the sequences of known proteins are stored, together with information on their biological functions, which facilitates such comparisons. "This kind of sequence analysis is a fundamental tool in the field of bioinformatics," explains Söding.
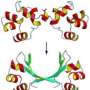
The sequence search programs assess sequence similarity by computing pairwise alignments: the two sequences of amino acids are arranged one above the other in such a way that mostly identical or similar amino acids are paired up in the same columns. "Perhaps even more important than the search for pairwise sequence similarities is the assembly of so-called multiple sequence alignments; in this case one searches for similar sequences in many related proteins and arranges them into a matrix, in which each sequence fills a row and similar amino acids end up in the same columns" says Söding. Because the functions and structure of evolutionarily related proteins are generally conserved - i.e. preserved even when the sequence is altered by mutations during the course of evolution - multiple sequence alignments form the basis for the prediction of the structure and molecular functions of uncharacterized proteins.
For the past 15 years, the program PSI-BLAST has been the most popular tool for the comparison of protein sequences, as it combines speed with high sensitivity and precision. Now Söding's team has designed a method, called HHblits, which clearly surpasses PSI-BLAST in all aspects of performance. This improvement is largely due to two factors. First the researchers convert both the sequence of interest and the sequences in the database to be searched into so-called Hidden Markov Models (HMMs). HMMs are statistical models that incorporate the mutation probabilities determined from sequence alignments – so this step increases the sensitivity and precision of the subsequent similarity search. In addition, the team has developed a filtering procedure that allows them to reduce the amount of data that needs to be searched without appreciable loss of sensitivity. The trick is first to assemble similar sequences from the database into multiple sequence alignments. Each alignment column is then labeled with one of 219 "letters", such that columns with similar amino acid composition are represented by the same letter. "By translating the multiple sequence alignments into sequences composed of these 219 letters, we can replace the time-consuming pairwise comparison of HMMs by the comparison of simple sequences", says Söding. This reduces the search time 2500-fold. Söding emphasizes that "HHblits allows to predict the function and structure of proteins more often and more accurately than was previously possible." His group is already working on further improvements to the method, for example by incorporating information on the three-dimensional structures of proteins.
More information: HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. M. Remmert, A. Biegert, A. Hauser, J. Söding. Nature Methods, 25.12.2011
Journal information: Nature Methods
Provided by Ludwig-Maximilians-Universitat Munchen