Calcium channel blockers caught in the act at atomic level

An atomic level analysis has revealed how two classes of calcium channel blockers, widely prescribed for heart disease patients, produce separate therapeutic effects through their actions at different sites on the calcium channel molecule.
Millions of Americans, and an even larger number of patients worldwide, take calcium channel blockers to control cardiovascular problems.
In a study published Aug. 24 in the advanced online edition of Nature, researchers describe how the fundamental mode of action of two distinct chemical classes of these drugs differs. The knowledge was gained by analyzing the atomic structure of their binding sites.
The paper is titled, "Structural basis for inhibition of voltage-gated Ca2+ channel by Ca2+ antagonist drugs."
UW Medicine researchers William A. Catterall, University of Washington professor and chair of pharmacology, and Ning Zheng, UW professor of pharmacology and investigator of the Howard Hughes Medical Institute, headed the project. The first author on the Nature report was Lin Tang, UW postdoctoral research scientist in pharmacology.
Calcium channel blockers were first identified and approved as medications in the previous half-century, and have emerged as major therapies for cardiovascular disorders. These latest findings could inform the design of new versions of calcium channel blockers that could be better and safer for managing heart beat irregularities, chest pain, and high blood pressure.
The researchers compared calcium channel blockers that treat heart arrhythmias, such as verapamil, with those taken for hypertension or angina, such as amlodipine.
The team wanted to learn how calcium channel blocker molecules interact with calcium channels, the molecular pores that govern the flow of calcium ions across a cell membrane. When these pores open in heart muscle cells or in the smooth muscle cells in arteries and veins, the rush of calcium entering the cells triggers contraction of the heart to pump blood and contraction of the arteries to narrow their diameter and thereby increase blood pressure.
By interfering with these molecular pores, calcium channel blockers can subdue a too-powerful cardiovascular response that can cause an irregular heart beat or high blood pressure.
Pharmacologists had thought that verapamil-like drugs physically blocked calcium channel pores to prevent calcium entry into the cell, and thereby restore a normal heart rhythm, whereas amlodipine-like drugs (called dihydropyridines) were thought to indirectly prevent calcium channel activation and pore opening, and thereby prevent high blood pressure and angina.
However, the specific structures behind these different mechanisms of action remained uncharted.
Advances in X-ray crystallography—a way to determine the arrangement of atoms within a large protein molecule—and in the functional analyses of ion channels have now enabled researchers to probe the sub-molecular depths of the drugs' actions. Researchers looked at where the drug molecules bound to calcium channels, and how this binding changes the workings of the channels.
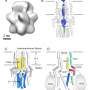
The binding site for the blood pressure and angina medication, amlodipine, was discovered to be on the outside edge of the calcium channel molecule. The central pore has voltage sensors around it which are sensitive to electrical potential.
The binding site is positioned on the outside edge of the central pore structure between two of the four subunits of the calcium channel molecule. The binding shuts down the channel by distorting its shape and lodging a calcium ion permanently within it.
 is a blood pressure medicine that remodels the channel from the outside, so that the calcium (red dots in center of image) lodges inside. Credit: Catterall and Zheng Labs/University of Washington")
"The amlodipine subtly remodels the pore so that the calcium ion is pulled to one side and just sticks there the whole time, as if it were locked up," said Ning Zheng.
In contrast, the verapamil molecule plugs the central cavity of a calcium channel and by itself directly barricades the calcium ion-conducting pathway.
Verapamil also takes advantage of the frequent openings of the calcium channels when the heart is palpitating, as it does during atrial fibrillation or atrial flutter. The more often the pore opens, the greater the odds that the verapamil molecule can slip into the central cavity and seal off the pore.
"Verapamil appears to bind better to calcium channels in the rapidly beating parts of the heart and slows them down," Catterall said.
He noted that his UW colleague, Bertil Hille, professor of physiology and biophysics, and his associates earlier had demonstrated the effects of rapid firing frequency on sodium channel blockade in their studies of local anesthetic drugs like lidocaine that prevent pain in dentistry and surgery.
On the other hand, in their resting state, calcium channels in blood vessel cells are usually closed. Amlodipine molecules modulate the voltage-dependent activation of calcium channels, and do not need to rely on frequent openings of the channel to enter the pore.
That is why amlodipine-like drugs, which relax the blood vessels, can treat certain causes of high blood pressure and the tight, squeezing pain of angina without major effects on the heart itself. This sets them apart from the verapamil-like drugs, which favor calcium channels in cells active in the electrical circuitry of the heart.
Understanding details of the two binding sites might lead to the development of calcium channel blockers that fit more exactly in place. A more precise shape might also prevent next-generation versions of the blockers from inadvertently aligning with the wrong binding sites and causing unwanted side effects.
"Calcium channel blockers are relatively safe drugs," Catterall explained, "but toxicity can arise from overdoses that can lead to ventricular arrhythmias or to too strong depression of the contraction of the heart or smooth muscle cells."
Structure-based, improved drug design, he said, might allow for smaller, yet still effective, drug doses that are more specific and safer. Fine-tuning the drug design, he added, might prevent another possible contributor to unwanted side effects: the off-target blocking of sodium channels by calcium channel blockers.
The new research was conducted on bacterial ion channels, which are the ancestors of sodium and calcium channels in other life forms, including worms, flies, fish and humans.
"These ancient channels in bacteria still recognize the drugs designed for people," Catterall said. "The experiments done by our second author Tamer Gamal El-Din, acting assistant professor of pharmacology, showed that these drugs act in the same way on bacterial channels as they do on those in mammals. It's remarkable that the most basic bacterial channels respond to these modern medicines for treating arrhythmias and certain other cardiovascular diseases. "
The study benefited from the federally sponsored Advanced Light Source at the Lawrence Berkeley National Laboratory, where the minute structures of the bound and unbound calcium channels could be analyzed.
Amazingly, Zheng explained that the experimental procedures carried out at the synchrotron beam line in California could be managed long-distance from the UW in Seattle via a simple laptop computer operated by Lin Tang and other investigators in this study.
More information: Lin Tang et al, Structural basis for inhibition of a voltage-gated Ca2+ channel by Ca2+ antagonist drugs, Nature (2016). DOI: 10.1038/nature19102
Journal information: Nature
Provided by University of Washington