Sequencing method precise enough to reveal mechanisms by which bacteria resist antibiotics
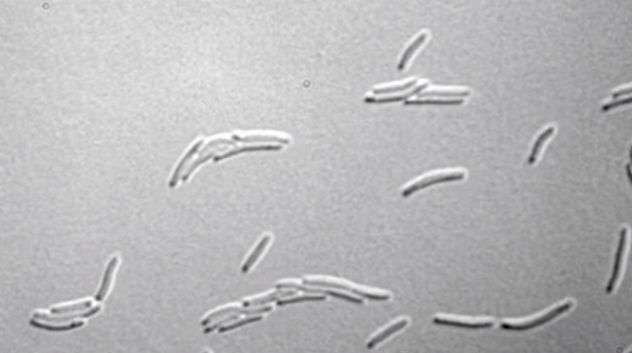
A new technology can read the order (sequence) of the "letters" making up DNA code with enough accuracy to reveal how bacteria use high-speed evolution to defeat antibiotics. That is the finding of a study led by researchers at NYU Langone Medical Center and published June 22 in the journal Nature.
The technology, called Maximum Depth Sequencing (MDS), eliminates the error introduced by core methods behind current high-speed DNA sequencing machines to catch genetic changes so rare that older methods could not tell them apart from machine error.
"We were able to directly measure for the first time both the standard change rate in DNA sequences across a bacterial genetic code, and the "hotspots" where bugs turn on genetic change many times faster than average to render antibiotics obsolete," says senior study author Evgeny Nudler, PhD, the Julie Wilson Anderson Professor of Biochemistry in the Department of Biochemistry and Molecular Pharmacology at NYU Langone.
"Beyond antibiotic resistance, the technology may soon give us the ability to find extremely rare genetic changes in any cell population, including cells in the bloodstream poised to become cancerous, and long before they seed tumors," says Nudler, also an investigator with the Howard Hughes Medical Institute.
Exceptionally Deep Sequencing
Advanced, high-throughput sequencing machines determine the order of the three billion bases making up a person's entire genetic code (genome) in about ten hours, with smaller, bacterial genomes taking less time. With this capability has come new understanding of how changes occurring randomly in the order of bases are linked to disease.
To determine the order of bases in a DNA sample, such technologies break DNA chains into pieces and use the enzyme DNA polymerase to make a copy of each fragment's sequence attached to a bar code, a tag that uniquely identifies each original DNA fragment. The machines then make a vast number of copies of each copy, enough to be picked up by technologies that use glowing probes to identify each letter in order.
The problem with standard methods is that any errors made in the original polymerase copying step show up in the all the copies, says Nudler. This leaves no way to tell errors apart from rare, naturally occurring changes in DNA sequence (mutations) increasingly linked to disease risk.
The first innovation described in the newly published paper was the use of polymerase to build bar codes off the ends of the original DNA fragments, rather than to make an error-prone copy of the fragment to be sequenced, and then amplifying the error. The method then makes multiple, independent copies of the barcoded original DNA fragments. In this way, any errors introduced by polymerase, or by the sequencing process in a given machine, show up in a few versions of the sequence generated, but not all in the same place, enabling their dismissal.
Rather than sequencing an entire genome, the new method zeroes in on much shorter DNA regions. By focusing the machine's capacity on strategic DNA "regions of interest," researchers were able to sequence each original fragment multiple times in a single run.
The study results revolve around the race between continual damage done to DNA chains by their environments and rapid DNA repair mechanisms. Experts estimate that DNA is damaged thousands of times an hour in a bacterial cell, but repair mechanisms mean that their DNA codes change slowly over time.
Using the new technology, the authors were able to observe mutations with enough statistical rigor to accurately calculate the standard, ongoing mutation rate in the bacterial species E. coli for the first time. Knowing the basal mutation rate also revealed to researchers the mutations occurring ten times more frequently than average in parts of the E. coli genome when exposed to antibiotics.
Specifically, the team found that doses of ampicillin and norfloxacin not large enough to kill bacteria outright, by causing oxidative stress and DNA damage in bacterial cells, turned down mismatch DNA repair - a system for repairing mistakes made as DNA is copied. Upticks in stress enabled bacteria to change their DNA code more quickly with the goal of evolving around treatments.
"We never would have seen these processes, but can now hope to harness them to take away a fundamental mechanism used by bacteria to acquire resistance," says Nudler.
Beyond the method's ability to find rare mutations in bacterial DNA, MDS promises to be useful in detecting rare mutants in human cell populations. It could conceivably identify rare "pre-cancer" genetic mutations in cells long before a tumor forms using a blood test, says Nudler. Related studies are already underway.
Along with Nudler, study authors were lead author Justin Jee, Aviram Rasouly, Ilya Shamovsky, Yonatan Akivis, Susan Steinman in the Department of Biochemistry and Molecular Pharmacology at NYU Langone, as well as Bud Mishra of the Courant Institute of Mathematical Sciences at New York University. This work was supported by the National Institute of Health (grant R01 GM107329), the Howard Hughes Medical Institute, and Russian philanthropist Timur Artemyev.
More information: Rates and mechanisms of bacterial mutagenesis from maximum-depth sequencing, Nature, DOI: 10.1038/nature18313
Journal information: Nature
Provided by New York University School of Medicine