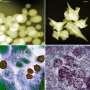
Mapping a cell's destiny: New tool speeds discovery of spatial patterns in gene networks
. Shown are a few of the 21 different “principal patterns” (PP), or pre-organ regions, they mapped. The principal patterns correspond to major organ systems in an adult fly (bottom). Credit: Siqi Wu, UC Berkeley and Berkeley Lab")
As in real estate, a key factor in gene expression and interaction is location, location, location. The same genes in different regions of a body may perform totally different functions. Determining the fate of these genes involves systematically mapping and detecting their spatial expression patterns, an unwieldy challenge because of the reams of data researchers must process.
It is a problem that researchers at the U.S. Department of Energy's Lawrence Berkeley National Laboratory and UC Berkeley are tackling with a new statistical method for extracting meaningful information from spatial gene expression data.
"We're investigating the process of development, of how the undifferentiated fertilized egg grows into a fully formed organism such as a fly or human being," said Erwin Frise, a Berkeley Lab scientist in the Division of Environmental Genomics and Systems Biology.
The challenge is born out of the success of genome sequencing – and the large volumes of RNA data subsequently generated – in recent decades. Frise and Berkeley Lab senior scientist Susan Celniker, in particular, have been working with the fruit fly genome as core members of the Berkeley Drosophila Genome Project, which Celniker co-directs.
Since the successful sequencing of the Drosophila melanogaster genome in 2000, the researchers have amassed large-scale collection of gene expression data covering the stages of a fruit fly's life, from egg to fully developed larvae.
"Our datasets are large and complex, requiring new methods to extract useful biological information about the regulatory networks that direct genes to differentiate into particular cells and tissue," said Celniker. "We determined spatial gene expression patterns, gene by gene, and now the goal is to identify the organizing principles."
The Berkeley Lab researchers teamed up with colleagues at UC Berkeley's Department of Statistics to tackle this challenge. Bin Yu, a professor of statistics, and her graduate student, Siqi Wu, worked with Frise to develop a learning algorithm that partitions biological spatial data or images into building blocks the researchers called principal patterns.
"These principal patterns correspond to pre-organ regions, areas that will become the different parts of the body," said Yu. "Genes work together to develop these pre-ordered regions."
They applied this algorithm to data obtained from fruit fly embryos only 1 to 3 hours old. Notably, in the development of organisms, cell fates are determined before structural features are visible.
The algorithm detected signs of related gene functions in the data to create a topological map of genetic regions destined to become the brain, midgut, mesoderm and more. The map contains 21 different principal patterns. They confirmed the results by matching them with a traditional fate map.
The method, described in a recent study, could go far in interrogating human tissue organization and helping elucidate key aspects of development, human health and disease.
More information: Siqi Wu et al. Stability-driven nonnegative matrix factorization to interpret spatial gene expression and build local gene networks, Proceedings of the National Academy of Sciences (2016). DOI: 10.1073/pnas.1521171113
Journal information: Proceedings of the National Academy of Sciences
Provided by Lawrence Berkeley National Laboratory