Viral gene editing system corrects genetic liver disease in newborn mice

For the first time, researchers have treated an animal model of a genetic disorder using a viral vector to deliver genome-editing components in which the disease- causing mutation has been corrected. Delivery of the vector to newborn mice improved their survival while treatment of adult animals, unexpectedly, made them worse, according to a new study by investigators from the Perelman School of Medicine at the University of Pennsylvania The team published their findings this week in Nature Biotechnology.
"Correcting a disease-causing mutation following birth in this animal model brings us one step closer to realizing the potential of personalized medicine," said senior author James Wilson, MD, PhD, a professor of Medicine and director of the Orphan Disease Center at Penn. "Nevertheless, my 35-year career in gene therapy has taught me how difficult translating mouse studies to successful human treatments can be. From this study, we are now adjusting the gene-editing system in the next phases of our investigation to address the unforeseen complications seen in adult animals." Wilson is also director of the Penn Gene Therapy Program.
The Wilson lab focused on liver as a target for gene editing since they had solved the problem of gene delivery in this organ in previous work using traditional gene therapy using vectors based on adeno-associated virus (AAV). However, gene replacement therapy with AAV is not ideal for treating genetic diseases of the liver that manifest as newborns since the non-integrating genome is lost as developing liver cells proliferate.
Because of this Wilson, co-first author Lili Wang, PhD, a research associate professor of Pathology and Laboratory Medicine, and collaborators, thought that the newborn liver might be an ideal organ for AAV-mediated gene correction using CRISPR-Cas9, an RNA-guided genome-editing technology that uses the bacteria protein Cas9. With CRISPR-Cas9 the corrected mutation will persist as the vector genome is lost.
This hypothesis was tested in a mouse model of a rare metabolic urea-cycle disorder caused by a deficiency in an enzyme called ornithine transcarbamylase (OTC). The urea cycle is a series of six liver enzymes that help rid the body of ammonia, a breakdown product of protein metabolism. When one of these enzymes is missing or deficient, ammonia accumulates in the blood and travels to the brain, causing a multitude of problems, including brain damage and death.
OTC deficiency is the most common of the urea-cycle disorders, occurring in one out of every 40,000 births. A mutated OTC gene can cause an enzyme that is shorter than normal, the wrong shape, or may not be produced at all. The genetic mutation responsible for OTC occurs on the X chromosome, so women are typically carriers, while their sons with the mutated gene suffer the disease.
Cut-and-Paste
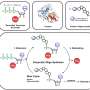
The team injected two AAVs (specifically an AAV8 serotype discovered in the Wilson lab that has an affinity for liver cells), one expressing Cas9 and the other expressing a guide RNA and a donor DNA, into newborn mice with OTC deficiency.
One AAV ferried the Cas9 enzyme via a liver-specific promoter to ensure it only expresses in liver cells when injected into the blood. The other AAV in the dual system ferried a guide RNA - a 20-base string of genetic building blocks followed by another sequence to lead the Cas9 enzyme to the correct spot within the DNA in the nucleus of the liver cell. The second AAV also contained a donor DNA template to correct the mutation so that the normal OTC protein can be made by the cell. The addition of this donor DNA to actually correct a mutation distinguishes this study from other recent genome-editing research findings that circumvent a mutation by deleting a portion of the normal gene.
This whole correction system is basically a "Cut-and-Paste" function, with the last part of the "Paste" phase relying on the cells' own DNA repair mechanism to properly join the OTC gene back together again.
In the newborn mice, injection of the AAV system reverted the mutation in 10 percent of liver cells, on average, as measured by the presence of the OTC enzyme in liver cells. They also saw an increased survival in young mice challenged with a high-protein diet, which makes OTC-deficient symptoms worse in the mice.
In contrast, more than 30 percent of the untreated OTC-deficient mice died after a week and their ammonia levels were significantly higher than the OTC mice whose genes were corrected. Deep sequencing of DNA isolated from liver cells in the treated mice also showed that correction to the mutation was consistent with the survival results.
On the other hand, gene correction in adult, eight-to-ten-week-old OTC-deficient mice was lower using the same dual-AAV system. The adults also showed diminished protein tolerance and lethal hyperammonia on a normal chow diet. After three weeks, the adult mice on a low dose of the gene correction started to die, and counterintuitively, mice given a high dose started to die nine days after injection.
"We were surprised by these results, but after some further investigation we deciphered the mechanism by which gene editing worsened the condition of the adult animals," Wang said. Looking at the DNA sequence in liver cells in adult mice, they found that the frequency of cells that had a corrected Paste function was only about one percent. "This was certainly not enough to help these adult mice," Wang noted. What was more problematic, and completely unexpected, is that many of the uncorrected genes contained large deletions that eliminated the residual activity of the mutant OTC gene.
The first step in correcting the gene is the creation of a break in the DNA by Cas9 in proximity to the mutation (the Cut) which, in the presence of the donor DNA, sets the stage for correction of the mutation in what is termed homology directed repair (HDR or the Paste). "It appears that HDR is more efficient in newborn liver cells than in adult liver cells." Wilson said.
In the absence of HDR the cell will repair the cut using another process called non-homologous end joining (NHEJ) that leaves in its wake small insertions or deletions. The team directed the cut to a part of the OTC gene that, if perturbed by a small insertion or deletion, would not interfere with the residual function of the mutant OTC gene. This was the case in newborn mice.
The team learned, however, that NHEJ in adult liver cells resulted in much larger deletions, some of which eliminated any residual function of the OTC gene. The net result of low rates of the Paste with responses to the Cut that destroyed the remaining gene function in many cells resulted in lower tolerance to protein in adult mice.
"The ontoward consequences of gene editing observed in adult OTC mice is limited to treating genetic diseases in which the mutation diminishes but does not eliminate function," Wilson explained. In an attempt to avoid this problem in certain adult patients with liver diseases, the team is exploring methods to create the Cut without inciting the large deletions while at the same time, driving higher frequencies of the Paste.
More information: A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice, Nature Biotechnology, DOI: 10.1038/nbt.3469
Journal information: Nature Biotechnology
Provided by University of Pennsylvania School of Medicine