Researchers define role of Tmem231 in maintaining ciliary function
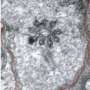
 localizes to the transition zone between the basal body (red) and axoneme of cilia (blue) in control cells (left) but is absent in cells from the mutant mice developed by Roberson et al (right). Credit: Roberson et al., 2015")
Researchers reveal how a protein linked to Meckel syndrome (MKS) and other human diseases regulates the membrane composition of cilia, finger-like projections on the surface of cells that communicate signals. The study appears in The Journal of Cell Biology.
MKS is a rare genetic disease characterized by kidney cysts, the presence of extra fingers and toes, and defects affecting several other organs. It is part of a class of disorders known as ciliopathies, meaning that it results from defects in the structure or function of cilia.
A multiprotein complex called the MKS complex assembles at a region of the cilium known as the transition zone. Mutations in some of the genes that encode MKS complex proteins disrupt ciliary membrane composition and cause MKS. A transmembrane protein called Tmem231 binds to the MKS complex protein B9d1, but how Tmem231 contributes to the assembly and function of the complex is unclear.
To investigate the role of Tmem231, a team of researchers led by Jeremy Reiter from the University of California, San Francisco, found that mice lacking the protein displayed MKS-like symptoms. MKS complex components, including B9d1, failed to localize at the ciliary transition zone in embryos from these mice, resulting in the loss of key signaling proteins from the ciliary membrane. These scientists additionally showed that the roundworm homologue of Tmem231 localizes to and controls transition zone formation and function, suggesting an evolutionarily conserved role for this protein.
Reiter and colleagues further studied the role of TMEM231 in humans. They were able to identify several additional mutations in the human TMEM231 gene, not only in MKS patients but also in two siblings with a ciliopathy called orofaciodigital syndrome type 3. All of these mutations were found to disrupt the MKS complex's organization and function.
More information: Roberson, E.C., et al. 2015. J. Cell Biol. DOI: 10.1083/jcb.201411087
Journal information: Journal of Cell Biology
Provided by Rockefeller University Press