Worm research defines the role of multiple disease genes in cilia
(PhysOrg.com) -- University College Dublin researchers led by Conway Fellow, Dr Oliver Blacque have outlined how cilia disease gene products regulate important aspects of early cilium formation and the integrity of the ciliary transport gate.
The findings from the international collaboration jointly led by University College Dublin, University of Alabama and Simon Fraser University, British Columbia are published in the current issuue of the Journal of Cell Biology.
Cilia are present on nearly all of our cells and act like cellular antennae. They play fundamental roles in many motility and sensory functions, including signalling pathways critical to development. Genetic mutations that disrupt the cilium structure and function are associated with a range of human diseases and syndromes, collectively called ciliopathies.
This research project used the nematode model, C. elegans, to focus on the functions of genes associated with two particular ciliopathies, Meckel syndrome (MKS) and Nephronophthisis (NPHP).
Describing the key findings, Dr. Oliver Blacque, co-senior author on this study said, “In worms, all cilia extend from the tips of dendrite structures on sensory neurons. We identified 7 MKS and MKS-like proteins, and 2 NPHP proteins that all localise to a specific ciliary sub-compartment at the base of C. elegans sensory cilia called the transition zone.
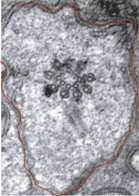
Using electron microscopy, we demonstrated that combinations of MKS and NPHP proteins, but not either protein alone, are required to properly dock cilia and the transition zone at the dendrite surface. This implies these disease-associated components play a key role at an early stage of cilium formation.
Furthermore, we demonstrated that proteins not usually found in cilia can abnormally enter the structure when MKS and NPHP genes are disrupted. It implies that these genes help to establish a gate keeping function for the transition zone at the base of cilia.”
These findings are significant because, for the first time, they define common novel functions for multiple MKS and NPHP proteins at the ciliary transition zone; a structure common to all ciliary subtypes from unicellular protists to humans. The data also helps to explain why patients with mutations in two ciliary disease gene products frequently present with more severe symptoms.
Each of the three laboratories contributed approximately equal amounts of data to the study. The Blacque laboratory (and specifically, Katarzyna Kida) contributed all of the electron microscopy studies (6 different combinations of MKS and NPHP mutations); the Leroux laboratory contributed some of the localisation data and some cilium structure/function data on the mutants; the Yoder laboratory contributed some of the localisation data and the data on ciliary gate restrictions. Efforts are ongoing to discover new transition zone proteins with roles in early cilium biogenesis and ciliary gate formation.
More information: MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. Corey L. Williams, et al. J. Cell Biol. jcb.201012116 Published March 21, 2011
Provided by University College Dublin