Error-correction strategy allows precise measurement of transcriptome in single cells
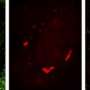
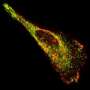
 for transcriptome imaging. Scheme at the top of the image: Schematic depiction of the identification of multiple RNA species using MERFISH, a single-molecule imaging method that uses combinatorial labeling and sequential imaging with encoding schemes capable of detection and/or correction of errors. RNA species (black lines) are encoded with a chosen set of N-bit binary words; during each round of imaging, only the subset of RNAs that should read 1 in the corresponding bit emit signals (yellow glow). The on/off sequence of fluorescent signals on each RNA molecule is translated into a binary code word of 1s and 0s, which is used to identify the RNA. Binary code words are chosen such that errors can be identified and in some cases corrected. Lower images: The detection of ~1000 different RNA species in a human fibroblast cell. Each spot in the main image on the left corresponds to an RNA molecule, colored according to its measured binary word. Inset: the composite, false-colored fluorescent image of the 14 hybridization rounds for the boxed sub-region with numbered circles indicating potential RNA molecules. White circles mark accurately decoded RNAs. Red circles mark RNAs decoded as errors. The fluorescent signals detected in individual hybridization rounds from this region are shown in the panels on the right, each false colored in a distinct hue. Credit: Science, April 9 2015")
Howard Hughes Medical Institute (HHMI) scientists have devised a method of visualizing RNA molecules inside cells so that the identity, location, and abundance of more than 1,000 different RNA species can be determined at the same time. The developers of the new technology say it should be possible to scale up the approach so that tens of thousands of RNA species can be imaged and identified in a single cell.
The approach, called multiplexed error-robust fluorescence in situ hybridization (MERFISH), is described in an article published online April 9, 2015, in Science Express. The technique uses combinatorial labeling, sequential imaging, and error-robust encoding schemes to identify numerous RNA species. In a specific implementation, the authors used tens of thousands of oligonucleotide probes that bind to cellular RNAs to encode each RNA species with a unique combination of readout sequences, and then used fluorescently labeled readout probes to detect these sequences during 14 or 16 rounds of hybridization. Unique combinations of readout probes bind to individual RNA molecules, spelling out a 14-bit or 16-bit code that identifies each one.
"We feel that we have worked out such a robust approach that we could feasibly scale up to the entire transcriptome [a cell's complete set of RNAs]," says Xiaowei Zhuang, an HHMI investigator at Harvard University who led the development of the new technique. "In this paper we report measurements of ~1,000 RNA species, already opening up many exciting applications, but there's no way that we're stopping here."
The core transcription machinery of RNA polymerase copies the information found in DNA genes onto messenger RNA molecules that then govern the production of proteins. The abundance of RNAs is commonly measured to indicate the relative activity of specific genes. Additional information can be gleaned by determining where inside a cell or tissue specific RNA molecules are located, Zhuang says, since the RNA location can influence where the encoded protein will perform its function.
An approach known as single-molecule fluorescence in situ hybridization (smFISH) has been valuable for imaging RNA molecules in their natural setting. smFISH, developed by Albert Einstein College of Medicine biologist Robert Singer, uses fluorescent probes made of DNA or RNA to detect specific sequences inside cells. Scientists can use it to quantify and determine the location of specific RNA molecules. By combining multiple probes for each RNA, the method has been used to simutaneously image up to ~30 different RNA molecules in individual cells.
"Single-molecule FISH has made enormous contributions to our understanding of cell biology," says Zhuang. But what if scientists could simultaneously image not just 30, but all 20,000 or so different protein-coding RNAs inside a single cell? Or all of those, plus RNAs that do not code for protein—bringing the total closer to 60,000?
Zhuang says she had been pondering about how to distinguish between such an overwhelming collection of molecules for a long time, and became increasingly more interested in this goal after seeing how much exciting knowledge has been learned from sequencing-based single-cell transcriptomic analyses. A biophycist who loves to invent novel imaging methods, Zhuang knew that imaging could offer the much needed spatial information of the transcriptome that is difficult for sequencing approaches to provide. She decided to make a new imaging tool for this purpose.
"Scientists can now synthesize a large collection of FISH probes–enough to bind selectively to each RNA within a cell. But it is not possible to visualize and discriminate between tens of thousands of fluorescent probes in a single image." Zhuang explains her thought process: "And imaging one at a time is similarly impractical. If we image one at a time, [to see all of the RNAs] you would have to image the same sample 20,000 or 60,000 times. That's just infeasible. Then it occurred to me that we can solve this problem using sequential imaging with combinatorial labeling to allow an almost unlimited number of patterns of probes to be associated with each RNA," Zhuang explains. "That would allow a massive number of RNA species to be identified."
Zhuang described her idea in her lab meeting and it was not a hard sell, she said. She and her team then devised special binary codes to encode individual RNAs, and labeling and imaging schemes to decode these RNA codes. After many rounds of experimental trial-and-error and more than two years of hard but exciting teamwork to improve accuracy and efficiency, the team is now unveiling their powerful imaging method.
First, they attached a set of "encoding probes" to RNA molecules in the cells. These probes bind specifically to target RNAs, encode them with a combination of readout sequence, and act as easily accessed landing sites for fluorescently labeled "readout probes" that are applied to the cells in subsequent rounds. After the first readout probe is added, an image is captured, revealing a fluorescent spot for each cellular RNA molecule that has bound readout probes. Those fluorescent spots are translated to the first bit of the binary code: any RNAs that fluoresce at this step are assigned a 1, whereas all other RNAs are assigned a 0. The process is repeated with a second readout probe, and each RNA that fluoresces is assigned a 1 for the second bit of the code. After 16 rounds, each RNA has a 16-bit string of 1s and 0s that uniquely identifies it. "In this way, you can image many RNAs in very few rounds," Zhuang explains.
Although 16 rounds of imaging could yield more than 60,000 unique binary codes, the team used only a special subset of these codes to encode their RNAs. That's because one major problem with spelling out a code over 16 rounds of imaging is that each round introduces a new opportunity for error. Even if the chance of misreading RNAs during a single round is low, the accumulated chance of error grows with the number of rounds. "In a code with 16 bits, if you use all of the more than 60,000 codes, a single error converts one RNA completely into another," Zhuang explains. "After 16 rounds of imaging, your accuracy becomes really lousy and most of what you have detected is wrong!"
To solve this problem, her team borrowed an error-correction strategy from the field of digital communications. Instead of assigning all possible codes as identifiers of specific RNAs, they use only codes that differ from all others by more than one bit. "In order to convert one valid word into another, you have to simultaneously make multiple errors in one RNA. That's much harder," Zhuang explains. Using codes that differ from all others by more than two bits even allows error correction. This limits the number of different RNAs that can be identified, but dramatically improves accuracy. "We image fewer RNAs by using these error robust codes, but that was a compromise that's well worth it," Zhuang says.
Singer, who is also a senior fellow at HHMI's Janelia Research Campus, calls Zhuang's approach a conceptual breakthrough. "It's made feasible something that a lot of us have been talking about trying to do, but we've been going at it a completely different way." By applying information theory to the problem, Singer says, Zhuang has gotten around limits to how many fluorescent labels can be discriminated as discrete colors. "We were all thinking you needed lots of fluorochromes, and we were limited by the spectral bandwidth. This is technically orders of magnitude beyond what we would have thought possible in the near future," he says.
Zhuang credits a talented interdisciplinary team – graduate student Kok Hao Chen and postdoctoral researchers Alistair Boettiger, Jeffrey Moffitt, and Siyuan Wang – with implementing a series of innovations that make MERFISH efficient and remarkably accurate.
Zhuang's team used MERFISH with an encoding scheme capable of both error detection and correction to simultaneously image 140 RNA species in individual human cells. They determined how many copies of each molecule were present, and found that their results closely matched the results of conventional smFISH measurements of several individual genes and, when averaged over hundreds of cells, also closely matched the results of bulk RNA sequencing measurements of all RNAs; both tried-and-true methods for quantifying specific RNA molecules. Using an alternative encoding scheme that detects but does not correct errors, they imaged a set of 1,001 RNA species. This scheme was less efficient at detecting RNAs, but still fairly accurately identified those that it detected. "Both schemes can be scaled up to image more RNA species," Zhuang says, but she thinks that the encoding schemes capable of both error detection and correction are more favorable when it comes the transcriptome-scale measurements because of their superior error-scaling property (slower increasing in error with the number of bits).
The ability to image hundreds to thousands of different RNA species in individual cells allows many interesting biological questions to be addressed. By examining cell-to-cell variation in the abundance of RNAs, Zhuang's team found patterns suggesting groups of genes that are commonly regulated. Their results allowed them to propose potential roles for about 100 genes of unknown function. They also observed interesting sub-cellular spatial patterns of RNA. The technique will also allow researchers to examine gene expression in individual cells without removing them from tissue, which could be particularly valuable in tissues like the brain, where elaborately structured cells are difficult to separate from one another.
More information: "Visualization of single RNA transcripts in situ." Science 24 April 1998: Vol. 280 no. 5363 pp. 585-590 DOI: 10.1126/science.280.5363.585
"Spatially resolved, highly multiplexed RNA profiling in single cells." zhuang.harvard.edu/Publication … hen_Science_2015.pdf
Journal information: Science Express , Science
Provided by Howard Hughes Medical Institute