A simple way to retrieve small genomes from a mix of various organisms

Scientists from the IZW led by Alex Greenwood publish in PLOS ONE a simple way to retrieve small genomes from a mix of various organisms.
Which viruses infect the elephant? Which type of bacteria causes severe lung disease in European brown hare? Molecular biological analyses of tissue samples always confront scientists with the same problem: how to retrieve the genome of a specific pathogen from a mixture of DNAs in a patient and its microbial cohabitants? "Very easily", says Alex Greenwood from the German Leibniz Institute for Zoo and Wildlife Research. "A short single-stranded base sequence is offered to the prepared DNA soup as a bait. Now, as it happens, not only does the complementary target sequence takes the bait, but by and by many other adjacent segments do so too." It does not even require a new method. The so-called "hybridisation capture method" offers everything that is needed. What is required is to pay attention during the subsequent data analysis.
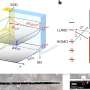
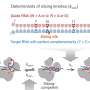
Greenwood's doctoral student Kyriakos Tsangaras discovered the additional value of hybridisation capture by chance. This technology is based on tiny magnetic beads with short baitsequences of a few base pairs (oligonucleotides, or oligos in short) attached to them. Once these prepared beads are added to a sample mix of single-stranded DNA fragments, only the target complementary sequences bind to the oligos and short double-stranded DNA fragments are created. The beads are removed from the sample with the help of a magnet and the loose fragments are rinsed off. Then, the short double-strands are eluted from the magnetic beads and sequenced.
On the day of discovery, Tsangaras only wanted to compare a particular sequence of DNA enclosed in the mitochondria of different southeast Asian rodents. He therefore used a sequence of about a thousand base pairs to capture the relevant DNA. "Yes, we have the sequence", he then told Greenwood. "But we also have much more!"
Analysis of the sequences and comparison with reference data demonstrated that the complete mitochondrial genome of the rodents had been retrieved from the "DNA pool". This does not make any sense at all, was Greenwoods first thought. However, control experiments led to the same intriguing result. Greenwood asked Tom Gilbert from the Center of GeoGenetics in Copenhagen to help analyse this phenomenon. After considering several hypotheses, they returned to the most obvious explanation– there must have been a chain reaction.
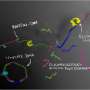
"Figuratively speaking the targeted sequence took the bait first – the complementary oligonucleotide sequence bound to the bait at the magnetic bead. Then a second sequence attached itself to the tail of the first, and the tail of the second then was 'bitten' by a third one and so on." Before being processed, the sample contained an intact double helix which then existed in fragments of various lengths. Because single-stranded DNA has the ability to spontaneously bind to any suitable complementary strand which it encounters, the following happened: After the complementary fragment from strand A bound to the bait, the flanking counterpart from strand B bound to its overhanging end. Then followed another fragment from A again, then from B, then A… and so forth.
It is quite simple and compatible with textbook knowledge. Why did not anyone else observe this before? "If someone is only looking for a thousand base pairs he usually only checks whether he found them. Everything that occurs in addition is discounted as junk", says Greenwood. The authors call this "by-catch" process, in which a single DNA fragment catches overlapping flanking sequences, "CapFlank". It is therefore possible to yield plenty of genetic information with just a tiny fragment. In fact, entire mitochondrial genomes and almost the entire genome sequence of a bacterium were obtained when specifically tested for the efficiency of the by-catch principle.
CapFlank opens doors to completely new possibilities, e.g. in the genetic analysis of pathogens. "We can use short preserved gene sequences to yield the genome (or at least large sections of it) from pathogenic variants of influenza viruses for example, or from completely new pathogens", explains Greenwood. As their next task, his team wants to retrieve simple and well characterised DNA viruses such as the elephant herpes virus.
The CapFlank-method is even suited for heavily fragmented "ancient DNA" extracted from animal bones from museum collections. These bones are often strongly contaminated with microbial or human DNA. Greenwood's colleagues successfully applied CapFlank to samples from koalas kept in museums. CapFlank is at its most efficient though with fresh DNA. From the intestinal bacterium Escherichia coli contained in a human urine sample the scientists retrieved 90 per cent of the genome in one go.
More information: Tsangaras K, Wales N, Sicheritz-Pontén T, Rasmussen S, Michaux J, Ishida Y, Morand S, Kampmann M, Gilbert MTP, Greenwood AD (2014): Hybridization capture using short PCR products enriches small genomes by capturing flanking sequences (CapFlank). PLOS ONE, PONE-D-14-23770R2 10.1371/journal.pone.0109101
Journal information: PLoS ONE
Provided by Forschungsverbund Berlin e.V. (FVB)