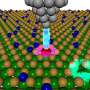
Physicists Use Computer Models to Reveal Quantum Effects in Biological Oxygen Transport
(Phys.org) —Physicists have created a unique combination of computer models, based on the theory of quantum mechanics, and applied them to a previously well characterised protein found in muscle to develop a new picture of how biomolecules transport and store oxygen (O2). In doing so, the international team have shown how the process of respiration, which is fundamental in humans and other vertebrates, exploits quantum mechanical effects working on tiny scales.
The physicists' discovery, building on a number of years of intense collaboration on theory and software development, has solved a long-standing problem at the interface of chemistry and biology. At the same time, they have demonstrated a new way by which quantum mechanics can be used to answer biochemical questions, with implications for inspiring drug-related research and further interdisciplinary collaborations.
Assistant Professor in Physics in the School of Physics at Trinity College Dublin, Dr David O'Regan, said: "This work helps to illustrate the fact that quantum mechanical effects, which may sometimes be viewed as somehow very exotic or only relevant under extreme conditions, are at play in the day-to-day regimes where biology, chemistry and materials science operate."
Iron-containing proteins, such as 'myoglobin' play a central role in biochemistry. Their ability to reversibly bind small molecules (such as O2) is vital for life. All animals must transport such molecules through the blood stream to where they are needed around their bodies, and myoglobin is particularly important in vertebrates. These proteins can also bind to other simple molecules such as carbon monoxide (CO), however. This is very dangerous in the case of the iron-containing proteins involved in respiration, since such a union is irreversible and leads to the protein being poisoned and the individual ultimately asphyxiating.
Until now, computational scientists have been unable to come to a good understanding of exactly why such protein poisoning is not more common. More specifically, computer simulations using the most widely-used theoretical approach (density-functional theory, 'DFT') that won the Nobel Prize for Chemistry in 1998, as well as many of its more advanced extensions, consistently predict that CO should bind to myoglobin much more readily than O2 when the two molecules are both present. If this was to happen in reality, we would not even be around to wonder why.
The mismatch between previous predictions and what we observe in nature prompted the team of physicists to develop a new approach to understand the process of how myoglobin preferentially binds to O2, and not CO. They combined their expertise in simulating both large systems and advanced approximations in quantum physics to reach their goal.
Dr O'Regan and his international collaborators used a special variety of DFT that is optimised for large systems (and on which Dr O'Regan has worked for a number of years) to model a large myoglobin structure. They also used another advanced approach, targeting the all-important iron atom, to treat some of the more complex interactions between its electrons.
It turns out that some electrons in the myoglobin involved in binding CO and O2 exhibit a strong 'entanglement' effect, which means that their motion cannot be described independently. The all-important strength of this effect is primarily controlled by a property of quantum mechanics (Hund's exchange) that has been traditionally neglected in such simulations; the team now believe that classical electric repulsion effects are far less important in determining which of CO and O2 is more energetically favourable for binding.
Dr O'Regan said: "We have succeeded in showing that quantum mechanical effects that we more often think of arising in advanced technological materials can be critical in determining the energy differences that drive biochemical processes occurring in the body. It is remarkable that myoglobin seems to be extremely well adapted to exploit the specific Hund's exchange strength of atomic iron, an intrinsically quantum mechanical property, in order to strongly promote O2 binding at the expense of CO. It is interesting, perhaps, to take a step back and even think of the implications with regards to early natural selection."
He added: "Computer-based simulation using DFT, together with its extensions developed in this work, is a laboratory for studying these effects on atomic length-scales. Approaches such as these are becoming increasingly valuable, and widely used, in helping to tackle contemporary, even urgent problems in areas such as pharmacology, materials for energy storage and conversion, and nanotechnology."
The team's findings were recently published in the prestigious peer-reviewed journal Proceedings of the National Academy of Sciences. The full article can be downloaded here.
Journal information: Proceedings of the National Academy of Sciences
Provided by Trinity College Dublin







.jpg)