Mapping activation of zygotic genome at high resolution
Embryonic development is initially controlled by maternal genetic information stored in the egg. LMU researchers now describe a methodology that allows the succeeding activation of the zygotic genome to be mapped at high resolution.
In multicellular organisms, the earliest steps in embryonic development, which are triggered upon fertilization of the egg, are regulated by RNA molecules transcribed during oogenesis and deposited in the egg cytoplasm. Subsequent activation of the genome of the embryo is accompanied by the progressive destruction of the maternal RNAs. During this latter phase, which is referred to as the maternal-to-embryonic transition (MET), the embryo contains a mixture of maternal RNAs inherited from the egg cell and RNAs expressed from its own zygotic genome formed by the fusion of the two haploid parental nuclei. "This makes it difficult to determine the precise timepoint and course of embryonic genome activation," says Professor Eckhard Wolf, who holds the Chair of Molecular Animal Breeding and Biotechnology at the LMU's Gene Center.
In collaboration with biochemist Dr. Helmut Blum of the Laboratory for Functional Genome Analysis at the Gene Center, Wolf's group has developed and tested a number of approaches which together enable the pattern of activation of the embryo's genome to be studied in detail. They report the results of their investigations in the latest issue of the Proceedings of the National Academy of Sciences (PNAS).
Timing the activation of 8000 genes
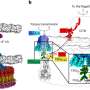
They chose early bovine embryos obtained by in vitro fertilization as a model system for their study. The embryos were derived from a cross between two subspecies of cattle, whose genome sequences differ sufficiently to allow transcripts originating from corresponding gene loci ("alleles") on the paternal and maternal chromosomes to be distinguished. The RNA present in the embryos at various times after fertilization was converted enzymatically into so-called cDNA, which was then analyzed using a high-throughput sequencing platform. This experimental design permitted the researchers to differentiate maternal RNAs stored in the egg from RNAs transcribed from the paternal alleles in the genome of the embryo itself.
The distinction is based on the presence of so-called Single Nucleotide Polymorphisms (SNPs), localized differences between the nucleotide bases present at corresponding positions in the parental genomes inherited by the embryo. By focusing on transcripts that encoded the paternal version of any given sequence, the researchers could determine when and in what temporal order the genes of the embryo were activated.
Most newly transcribed RNAs undergo a series of processing steps, which remove segments of the primary sequence, before the mature RNAs can leave the cell nucleus. So Wolf and his colleagues could also use their sequence data to discriminate between the mature forms of maternal RNA present in the egg cytoplasm and newly synthesized immature RNAs that had not yet left the nucleus. "These two complementary strategies allowed us to determine the timepoints of activation of nearly 8000 zygotic genes during early bovine development," Wolf explains.
The data reported by Blum, Wolf and their colleagues demonstrate how a combination of biotechnological methods and state-of-the-art sequencing procedures can yield new insights into the regulation of early mammalian development. The findings are of particular interest in the context of assisted reproduction techniques, and in cases where the mother suffers from a specific metabolic disorder.
Journal information: Proceedings of the National Academy of Sciences
Provided by Ludwig Maximilian University of Munich