Mitochondrial cooperatives
Mitochondria, the organelles that supply the cell with energy, are highly dynamic and can link up to form complex tubular networks. A new study shows that this response can transiently compensate for a shortfall in energy production.
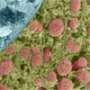
In animal cells, mitochondria produce most of the energy required for growth and proliferation, in the form of the universal unit of biological energy – ATP. Many other essential metabolic processes also take place in these membrane-bounded organelles, which have their own small genomes. They also play crucial roles in cellular aging and programmed cell death. In textbooks, they are often depicted as bean-shaped organelles. But in recent years it has become clear that mitochondrial morphology is highly dynamic. Indeed, individual mitochondria can undergo repeated fusion to form branched tubular networks, whose form varies depending on the cell type. Mitochondria in muscle cells, for example, link up to form long cables.
Cheese instead of cables
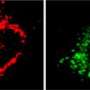
"Why mitochondria display so much variability in form and structure is one of the most exciting open questions in cell biology," says Barbara Conradt, Professor of Cell and Developmental Biology at LMU. Together with postdoc Stéphane Rolland and other members of her group, Conradt is trying to understand the functional significance of the morphological diversity of mitochondria. The team uses the nematode Caenorhabditis elegans as a model system, and their latest work focuses on a mutant in which muscle mitochondria fail to form the typical cables, owing to the partial inactivation of the nuclear gene mma-1. "Instead of linking up into well-defined cables, the mitochondria in the mutant overdo it, and undergo hyperfusion to form a large structure that looks rather like a chunk of Swiss cheese," says Conradt.
Loss of mma-1 also impairs the function of a protein complex involved in the synthesis of ATP in the mitochondria. Nevertheless, the researchers found, much to their surprise that mutant cells produce about the same amount of ATP as normal cells, despite making only half as much of the protein product encoded by the mma-1 gene. "We concluded from this that hyperfusion enhances the efficiency of ATP synthesis," says Conradt. This idea is supported by the finding that inhibition of hyperfusion renders the mma-1 mutant inviable.
Mitochondria and neurodegeneration
In collaboration with Konstanze Winklhofer, Conradt and her coworkers found the same hyperfusion phenotype in mammalian cells in which the mma-1 homolog, the gene LRPPRC, was partially inactivated. As in C. elegans, hyperfusion can transiently compensate for defects in ATP production. A few days after gene inactivation, energy production collapses in the mutant cells. "This is the first time that hyperfusion has been shown to represent an attempt to counteract the effects of a genetic defect in mitochondrial energy production. It probably works for only a limited time because the mutation involved also has an adverse impact on other mitochondrial functions," Conradt suggests.
Interestingly, mutations in the mammalian LRPPRC are associated with one form of Leigh syndrome, a serious neurodegenerative disorder that is characterized by a perturbation in mitochondrial energy metabolism. "So our findings are clearly of medical relevance, and could lead to new insights into this disease, as our C. elegans mutant provides an excellent experimental model for the study of its pathogenesis," says Conradt. She and her team will now test whether stimulating mitochondrial ATP production will reverse the hyperfusion phenotype. If sufficient numbers of functional mitochondria could be recovered in this way, the organism could perhaps compensate for defects in mma-1/LRPPRC in the long term.
More information: PNAS 2013. www.pnas.org/content/early/201 … /1303872110.abstract
Journal information: Proceedings of the National Academy of Sciences
Provided by Ludwig Maximilian University of Munich