Clustering is key to lighting up the dark proteome
 and often ignore Spectrum-Spectrum Matches (SSMs), especially if PSMs for these SSMs are not established. However, SSMs also are useful when the corresponding peptide is not identified, because they allow a researcher to cross-reference spectra generated by different researchers and to query all spectra ever generated against a single repository.
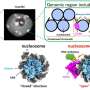
Spectral libraries are essentially databases of PSMs, while spectral archives are databases of both PSMs and SSMs. Although construction of PSMs (via tandem mass spectrometry database search) is a well-studied topic, construction of all SSMs represents a formidable clustering problem. The figure reveals similarities and highlights differences between construction (left) and use (right) of spectral libraries and spectral archives.
With an archive, researchers first cluster, then search the clusters against a protein database to generate Peptide-Cluster Matches (PCMs). In turn, these PCMs get propagated to all spectra in the identified clusters to generate PSMs. With the library, researchers first search the spectra against a protein database to generate PSMs, group PSMs corresponding to the same peptide, and finally deposit the curated consensus PSM in the spectral library. Then, the spectral library can be used to identify spectra from new spectral datasets.")
(PhysOrg.com) -- A new approach that organizes previously unused mass spectra from proteomics studies gives scientists the ability to use these spectra to gain more information about proteins in a wide range of organisms. Scientists from the University of California-San Diego and Pacific Northwest National Laboratory have created a vast spectral archive from more than a billion mass spectra acquired at PNNL between 2001 and 2009. They describe their approach in the July issue of Nature Methods.
In recent years, the volume of tandem mass spectrometry data generated from proteomics experiments has increased dramatically. Multiple, nearly identical mass spectra of the same peptides are routinely measured by various laboratories. Scientists compare the spectra with peptides residing in a database of known protein sequences. They then evaluate the resulting matches using various scoring methods to assign an identity to the peptide spectrum. Large sets of spectra can be organized into spectral libraries where other spectra can be brought for comparison, leading to increasing effectiveness in peptide assignments used for protein identifications.
But what about those spectra not identified; that is, those not associated with a known peptide? Typically, unidentified spectra are ignored or discarded, as they have limited value to the researchers because the protein is unidentified. As a result, a significant fraction of the proteins remain unidentified, constituting an effective "dark proteome" of unknown content.
Shedding light on the dark proteome is where the UCSD/PNNL team comes in. While spectral libraries discard unidentified spectra, spectral archives use all mass spectra—identified or unidentified-as clusters (see "Spectral Archives Complement Spectral Libraries"). The scientists not only showed the feasibility of constructing large archives and their basic utility for run-of-the-mill peptide identification, they developed new applications now possible because a diverse collection of datasets can be analyzed as a whole.
"We believe that spectral archives could change the nature of proteomics by motivating researchers who are analyzing seemingly unrelated data to share this data," said senior author Dr. Pavel Pevzner, UCSD. "Doing so improves the quality of the interpretations of both of their spectral datasets."
With archives, a researcher can identify clusters of spectra from different organisms. Besides indicating that such spectra are interesting—as they are likely to indicate proteins occurring over multiple species—this fact can be used to reduce the effective protein database size, leading to new, confident peptide and protein identifications. The team also showed that short peptides (shorter than 7 amino acids) could be confidently identified, which is much more difficult with typically used approaches.
The PNNL mass spectra data used by the team included samples taken from a diverse set of more than 100 organisms, including humans, the common house mouse, and the metal-reducing bacterium Shewanella oneidensis. The research team developed a clustering tool, MS-Cluster, that generated a spectral archive from the ~1.18 billion spectra from PNNL. This archive greatly exceeds the size of existing spectral repositories.
To evaluate whether spectral archives can increase peptide identifications, the researchers selected a subset of 14.5 million spectra from the microorganism S. oneidensis and constructed an archive with them. They did this by breaking the dataset into five sets of ~2.9 million spectra then incrementally adding each set of spectra to the archive. At each stage they compared the number of protein and unique peptide identifications made by searching the clusters in the archive with the number that could be obtained with conventional database search approaches.
The archive consistently yielded more unique peptide and protein identifications. With the archive, the scientists also were able to identify many more spectra through their cluster membership. At different stages, they identified 50-75% more spectra through cluster membership than via a regular database search.
This study also highlights the large number of spectra for which peptide and protein identifications are not achieved, opening the door for use of experimental and computational approaches to identify the significant numbers of peptides effectively ignored by proteomics studies to date.
More information: Frank AM, et al. 2011. "Spectral archives: extending spectral libraries to analyze both identified and unidentified spectra." Nature Methods 8(7):587-591. DOI:10.1038/nmeth.1609
Provided by Pacific Northwest National Laboratory