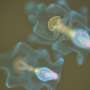
Parkinson's disease may be caused by microtubule, rather than mitochondrial complex I, dysfunction
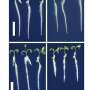
 contains the same number of tyrosine hydroxylase-positive dopaminergic neurons as wild-type mice (left). The study appears in the March 7 issue of the Journal of Cell Biology. Credit: Image credit: Choi, W.-S., et al. 2011. J. Cell Biol. doi:10.1083/jcb. 201009132")
Patients with Parkinson's disease (PD) suffer a specific loss of dopaminergic neurons from the midbrain region that controls motor function. The exact mechanism of this selective neurodegeneration is unclear, though many lines of evidence point to dysfunctional mitochondrial complex I as one root cause of the disease. Yet new research now suggests that defective regulation of microtubules may be responsible for at least some cases of PD. The study appears in the March 7 issue of The Journal of Cell Biology.
Mitochondria were first implicated in PD when drug users in California developed Parkinson's-like symptoms due to a contaminant in their heroin supply called MPTP. This chemical is metabolized in the brain into MPP+, a compound that blocks oxidative phosphorylation by inhibiting mitochondrial complex I. Subsequently, other chemicals such as rotenone were shown to inhibit complex I and induce PD in animal models, and PD patients were found to have reduced levels of complex I activity. Moreover, several proteins linked to genetic forms of PD are involved in maintaining normal mitochondrial function. But in 2008, Zhengui Xia and colleagues at the University of Washington in Seattle began to cast doubt on complex I's guilt.
"We didn't set out to prove anybody wrong," says Xia. "We just wanted to do a quick experiment to provide genetic proof that complex I inhibition causes dopaminergic neuron death." Xia and colleagues therefore examined mice lacking an essential subunit of complex I called Ndufs4. To their surprise, dopaminergic neurons from these mice were healthy and remained sensitive to MPP+- and rotenone-induced death.
This suggested that complex I inhibition is insufficient to cause dopaminergic nerve apoptosis. Confirming this, the team found that another complex I inhibitor, piericidin A, has no effect on dopaminergic cell survival. "So if it's not by inhibiting complex I, what is the mechanism [by which rotenone and other drugs kills these neurons]?" says Xia.
The researchers turned their attention to another property of rotenone: the drug also depolymerizes microtubules. Cultured dopaminergic neurons were spared from rotenone-induced death if they were co-treated with the microtubule-stabilizing drug taxol. On the other hand, the depolymerizing agent colchicine was as deadly as rotenone to dopamine-producing neurons.
But why do these microtubule-disrupting drugs only kill dopaminergic neurons and not other types of nerve cells? "Microtubule disassembly impairs dopamine release, so dopamine accumulates in the cell," explains Xia. Excess cytoplasmic dopamine may then be oxidized, producing reactive oxygen species (ROS) that induce cell death. Taxol prevented rotenone from increasing dopamine and ROS levels, and drugs that inhibit either dopamine synthesis or ROS protected neurons from rotenone-induced death.
Rotenone may have an additional dirty trick up its sleeve. The researchers think that the drug also inhibits VMAT2, the protein that transports dopamine into synaptic vesicles. Boosting VMAT2 expression promoted dopaminergic neuron survival, presumably by removing excess dopamine from the cytoplasm, thereby limiting the neurotransmitter's oxidation.
Yet mitochondrial complex I isn't a completely innocent bystander. Dopaminergic neurons lacking Ndufs4 are actually more sensitive to rotenone than wild-type neurons. The loss of complex I causes its substrate, NADH, to accumulate, which in turn boosts the activity of the enzyme that synthesizes dopamine. "The basal dopamine level is higher in knockout cells," explains Xia. "It's not high enough to cause a problem by itself, but it synergizes with rotenone's inhibition of microtubules and VMAT2."
Simultaneous inhibition of mitochondrial and microtubule function by different environmental and genetic factors may therefore contribute to PD. Indeed, several proteins linked to familial PD can influence the microtubule cytoskeleton. The E3 ubiquitin ligase Parkin, for example, targets alpha- and beta-tubulin, as well as dysfunctional mitochondria, for degradation. Xia says she now wants to confirm her group's findings in vivo. Ndufs4-knockout mice die at 7 weeks, so conditional knockout models will be needed to check for PD-like symptoms in older animals.
More information: Choi, W.-S., et al. 2011. J. Cell Biol. doi:10.1083/jcb.201009132
Provided by Rockefeller University