Semi-automated 'pathwalking' to build a protein model
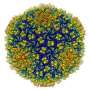
A new semi-automated tool called pathwalking makes it possible to generate a "first draft" model of a protein fold taken from near-atomic resolution images of between three and six angstroms (Å), said researchers at the National Center for Macromolecular Imaging in the department of biochemistry at Baylor College of Medicine.
In a report that appears online in the journal Structure, the BCM team describes the development of the semi-automated protocol that enables researchers to "rapidly generate an ensemble of initial models for individual proteins, which can later be optimized to produce full atomic models."
Taking the 3-D images generated through the process of electron cryo-microscopy and X-ray crystallography, the team developed this computational approach to produce these first-generation models of the proteins' structure or fold without prior knowledge of the protein's sequence or other information.
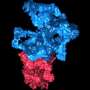
"This is important in working with big complexes made up of 10 to 30 proteins," said Dr. Matthew Baker, instructor in biochemistry and molecular biology at BCM and the paper's corresponding author. "You might know the structure of one or two proteins, but you want to know how all of those proteins interact with each other. As long as you can separate one protein from another, you can use this technique to make a model of each of the proteins in the complex."
"We borrowed from a classic computer science problem called the 'traveling salesman problem,'" said Dr. Mariah Baker, the paper's first author and a postdoctoral fellow at BCM. "It is in effect a connect-the-dots puzzle without the numbers."
In the traveling salesman problem, computer programmers are asked to figure the best route for a salesman who wants to visits all the cities where he sells just once while minimizing the distance traveled. Pathwalking solves a similar problem for proteins by looking for the optimal path through a 3-D image that connects C-alpha atoms, rather than cities, to form the protein's structure.
The tool is the answer to the dilemma presented by the near-atomic structures that are in the "middle" – not of the highest resolution or the lowest resolution, said Matthew Baker.
As many as 25 percent of all structures imaged by electron cryo-microscopy and one-third of large protein complexes solved by X-ray crystallography are in the 3 to 10 angstroms range, said Matthew Baker.
Until now, the methodology used to annotate or trace the structure of protein from these density maps was usually tailored to specific cases, said Mariah Baker.
"They involved a lot of user intervention and the possibility to include bias," she said. That sparked a determination to automate the process with better routines that required less specific information.
"The question we asked was, can we trace a protein fold in a density map without a priori knowledge," she said. "The answer is that we can."
Provided by Baylor College of Medicine