This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Machine learning tool simplifies one of the most widely used reactions in the pharmaceutical industry
. DOI: 10.1126/science.adg2114")
In the past two decades, the carbon-nitrogen bond forming reaction, known as the Buchwald-Hartwig reaction, has become one of the most widely used tools in organic synthesis, particularly in the pharmaceutical industry given the prevalence of nitrogen in natural products and pharmaceuticals.
This powerful reaction has revolutionized the way nitrogen-containing compounds are made in academic and industrial laboratories, but it requires lengthy, time-consuming experimentation to determine the best conditions for a highly effective reaction.
Now, Illinois researchers in collaboration with chemists at Hoffman La-Roche, a pharmaceutical company in Switzerland, have developed a machine learning tool that predicts in a matter of minutes the best conditions for a high-yielding reaction with no lengthy experimentation.
In a recently published article in Science, Illinois chemistry professor Scott Denmark and Ian Rinehart, a recent Ph.D. graduate in the Denmark lab, describe how they developed, trained, and tested their machine learning model to drastically accelerate the identification of substrate-adaptive conditions for this palladium–catalyzed carbon-nitrogen bond forming reaction.
Denmark said this reaction is a very general transformation so there is much structural diversity among reactant pairings and a lot of "levers to pull" to make it work.
"And that's what we have figured out," Denmark said.

User guides and cheat sheets have evolved in the nearly 30 years since this reaction was discovered, and they can provide some direction, Rinehart explained, but experimentation is often necessary. Basically, a trial-and-error process in a lab.
"It's a problem that everyone in the pharmaceutical industry recognized was ripe for intervention by informatics methods," Denmark said. "Lots of people have tried to use the US Patent and Trademark Office or Chemical Abstracts or other huge databases to try to model to make predictive tools for this one very important reaction. But they haven't been able to do very well because the information in the literature is just not very reliable."
The design and construction of their machine learning tool required the generation of an experimental dataset that explores a diverse network of reactant pairings across a set of reaction conditions. A large scope of C–N couplings was actively learned by neural network models by using a systematic process to design experiments.
The challenge for a project like this, Denmark said, was the amount of potential data to collect and the thousands and thousands of experiments required to build a database of information for modeling.
"One of Ian's biggest contributions was figuring out the workflow to decide what experiments to do to get a valid predictive model with about 3,500 experiments and still be able to make predictions without an enormous database," Denmark said.
They also experimentally validated the predictions from the machine learning tool.
"We tested them and found with pretty good statistics that the conditions were producing compounds when we expected," Denmark said.
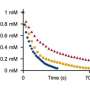
The researchers report that their models showed good performance in experimental validation: Ten products were isolated in more than 85% yield from a range of couplings with out-of-sample reactants designed to challenge the models.
Rinehart said they taught machine learning models to have a kind of chemical intuition like what an expert has.
"So, we have now run or talked about so many of these couplings that we have a good intuition about what's going to happen, but someone who hadn't run hundreds or thousands of these might not have a good first guess. We have taught a model at a much more granular level [than user guides] to have an intuition. It's not perfect. But that's kind of the point. It doesn't have to be. It just has to get you to the answer faster," Rinehart said.
And the coolest part, Rinehart explained, is that intuition gets honed over time as more people use the machine learning tool. The developed workflow continually improves the prediction capability of the tool as the corpus of data grows.
"It's an exciting time as data science merges with chemistry," Denmark said. "And this is the perfect marriage. A lot of people recognized this, but no one has done it, at least not in a meaningful way that is experimentally validated."
The Denmark group is creating a cloud-based version of the workflow to enable scientists around the world to use this tool which will continuously add data to improve the model as more structurally diverse substrates are tested and different catalysts and conditions are added to the database.
Rinehart said the code is public and on an open-source license, so anyone can download and use it. Also, he is currently working on a more user-friendly interface that will allow someone to draw the two molecules they want to react, copy and paste them into the program, and get predictions in minutes instead of hours, depending on the complexity of the molecules.
"I think it's really exciting to do something like that," Rinehart said. "We don't often publish a paper and put out a tool in the public domain that people can use in the field. People in academic labs like ours could use this tool and get an answer faster in their own research."
More information: N. Ian Rinehart et al, A machine-learning tool to predict substrate-adaptive conditions for Pd-catalyzed C–N couplings, Science (2023). DOI: 10.1126/science.adg2114
Journal information: Science
Provided by University of Illinois at Urbana-Champaign