This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Machine-learning-boosted drug discovery with 10-fold time reduction
. DOI: 10.1021/acs.jcim.3c01239")
Boosting virtual screening with machine learning allowed for a 10-fold time reduction in the processing of 1.56 billion drug-like molecules. Researchers from the University of Eastern Finland teamed up with industry and supercomputers to carry out one of the world's largest virtual drug screens.
In their efforts to find novel drug molecules, researchers often rely on fast computer-aided screening of large compound libraries to identify agents that can block a drug target. Such a target can, for instance, be an enzyme that enables a bacterium to withstand antibiotics or a virus to infect its host. The size of these collections of small organic molecules has seen a massive surge over the past years.
With libraries growing faster than the speed of the computers needed to process them, the screening of a modern billion-scale compound library against only a single drug target can take several months or years—even when using state-of-the-art supercomputers. Therefore, quite evidently, faster approaches are desperately needed.
In a study published in the Journal of Chemical Information and Modeling, Dr. Ina Pöhner and colleagues from the University of Eastern Finland's School of Pharmacy teamed up with the host organization of Finland's powerful supercomputers, CSC—IT Center for Science Ltd—and industrial collaborators from Orion Pharma to study the prospect of machine learning in the speed-up of giga-scale virtual screens.
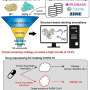
Before applying artificial intelligence to accelerate the screening, the researchers first established a baseline: In a virtual screening campaign of unprecedented size, 1.56 billion drug-like molecules were evaluated against two pharmacologically relevant targets over almost six months with the help of the supercomputers Mahti and Puhti, and molecular docking. Docking is a computational technique that fits the small molecules into a binding region of the target and computes a "docking score" to express how well they fit. This way, docking scores were first determined for all 1.56 billion molecules.
Next, the results were compared to a machine learning-boosted screen using HASTEN, a tool developed by Dr. Tuomo Kalliokoski from Orion Pharma, a co-author of the study.
"HASTEN uses machine learning to learn the properties of molecules and how those properties affect how well the compounds score. When presented with enough examples drawn from conventional docking, the machine learning model can predict docking scores for other compounds in the library much faster than the brute-force docking approach," Kalliokoski explains.
Indeed, with only 1% of the whole library docked and used as training data, the tool correctly identified 90% of the best-scoring compounds within less than 10 days.
The study represented the first rigorous comparison of a machine learning-boosted docking tool with a conventional docking baseline on the giga-scale. "We found the machine learning-boosted tool to reliably and repeatedly reproduce the majority of the top-scoring compounds identified by conventional docking in a significantly shortened time frame," Pöhner says.
"This project is an excellent example of collaboration between academia and industry, and how CSC can offer one of the best computational resources in the world. By combining our ideas, resources and technology, it was possible to reach our ambitious goals," said Professor Antti Poso, who leads the computational drug discovery group within the University of Eastern Finland's DrugTech Research Community.
Studies on a comparable scale remain elusive in most settings. Thus, the authors released large datasets generated as part of the study into the public domain. Their ready-to-use screening library for docking enables others to speed up their respective screening efforts, with 1.56 billion compound-docking results for two targets that can be used as benchmarking data.
This data will encourage the future development of tools to save time and resources and will ultimately advance the field of computational drug discovery.
More information: Toni Sivula et al, Machine Learning-Boosted Docking Enables the Efficient Structure-Based Virtual Screening of Giga-Scale Enumerated Chemical Libraries, Journal of Chemical Information and Modeling (2023). DOI: 10.1021/acs.jcim.3c01239
Journal information: Journal of Chemical Information and Modeling
Provided by University of Eastern Finland