This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
trusted source
proofread
MAGUS: Crystal structure predictions assisted by machine learning and graph theory

Crystal structures have a decisive impact on the properties of materials, and research on crystal structures often serves as a starting point for material studies. Crystal structure prediction is a method that seeks stable or metastable structures solely based on the chemical composition under specific conditions. It has become an effective tool for discovering new materials and exploring phase space.
Crystal structure prediction methods can assist in determining structures experimentally or designing specific structures to guide experimental synthesis, thereby significantly reducing experimental costs. Therefore, it holds great scientific significance.
However, current crystal structure prediction techniques are still limited because of two aspects: the cost of performing structure optimization using first-principles calculations increases cubically with the number of atoms, and the number of local minima on the potential energy surface increases exponentially with the increasing degrees of freedom.
To tackle the first issue, researchers propose the use of a machine learning potential for surrogate calculations. After generating random structures, a subset of these structures is randomly chosen for performing DFT single-point energy, force, and stress calculations to construct a training set.
The initial machine learning potential is then trained using this dataset. In the subsequent search process, the computationally expensive DFT structure optimizations are replaced with the machine learning force field. During the optimization, the structures that require extrapolation are recorded.
If the extrapolation exceeds a predetermined threshold, DFT single-point calculations are performed on those structures, and they are added to the training set for retraining the force field to correct the potential function. This process continues until the optimization process can be carried out entirely by the machine learning surrogate. Due to the errors introduced by the machine learning force field, calibration using first-principles calculations is necessary after the search concludes.
To address the second issue, the research team chose to utilize graph theory to reduce the search space. An important concept in evolutionary algorithms is to preserve excellent "genes" from the parent generation to the offspring generation. For crystal structures, these "genes" represent local structural motifs.
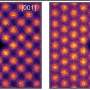
While these excellent motifs can be molecules in molecular crystal searches, it is not easy to determine them for ordinary extended crystals. However, by abstracting periodic crystal structures as graphs, community detection algorithms can be employed to accomplish this task. For example, in the case of an extended α-boron structure, when it is transformed into a quotient graph and subjected to community detection algorithms, the extracted 0-dimensional component corresponds to the boron icosahedron.
By keeping this component unchanged in subsequent genetic operations, the degrees of freedom decrease from 36 to 6, significantly reducing the search space.
In subsequent testing, the research team found that the machine learning approach could reduce the number of self-consistent calculations by approximately two orders of magnitude, while graph theory helped reduce the number of structures that needed to be visited by 30% to reach the target structure. By employing this method, the research team has achieved a series of breakthroughs in fields such as planetary science, super-hard materials, high-energy density materials, and superconducting materials.
The work is published in the journal National Science Review.
More information: Junjie Wang et al, MAGUS: machine learning and graph theory assisted universal structure searcher, National Science Review (2023). DOI: 10.1093/nsr/nwad128
The source code of MAGUS can be accessed after registration at: www.wjx.top/vm/m5eWS0X.aspx
Provided by Science China Press