This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
New method developed to infer gene regulatory networks from single-cell transcriptomic data
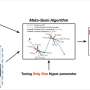
 Three types of datasets that can be dealt with the STGRNS. Data type 1 is scRNA-Seq data without pseudo-time ordered cells. Data type 2 is scRNA-seq with pseudo-time ordered cells. Data type 3 is time-course scRNA-seq data. (b) The training strategy for the GRN reconstruction. The same TFs and genes exist in the training and testing datasets. The GRNs reconstruction adopts this strategy. (c) The training strategy for the TF–gene prediction. The training dataset and the test dataset have the same genes but not the same TFs. We demonstrate one loop using threefold cross-validation. The size of each fold is not equal because the size of the TGs of each TF is different. The TF–gene prediction adopts this strategy. (d) The output of STGRNS for network inference. Credit: Bioinformatics (2023). DOI: 10.1093/bioinformatics/btad165")
Single-cell RNA-sequencing (scRNA-seq) technologies offer the opportunity to understand regulatory mechanisms at single-cell resolution. Gene regulatory networks (GRNs) provide a crucial blueprint of regulatory mechanisms in cellular systems and thus play a central role in biological research. It is therefore imperative to develop an accurate tool for inferring GRNs from scRNA-seq data.
Researchers from the Wuhan Botanical Garden of the Chinese Academy of Sciences have developed a novel method, namely STGRNS, for constructing GRNs from scRNA-seq data using a deep learning model. Results have been published in Bioinformatics and the tool and tutorial are publicly available at https://github.com/zhanglab-wbgcas/STGRNS.
In this algorithm, a gene expression motif technique was proposed to convert each gene pair into a form that can be received as a transformer encoder. By avoiding missing phase-specific regulations in a network, STGRNS can accurately infer GRNs from static, pseudo-time, or time series single-cell transcriptome data.
The researchers showed that STGRNS outperforms other state-of-the-art deep learning methods on 48 benchmark datasets, including 21 static scRNA-seq datasets and 27 time-series scRNA-seq datasets.
Unlike other "black box" deep learning-based methods, which are often characterized by their opacity and the associated difficulty in providing clear justifications for their predictions, STGRNS is more reliable and can interpret the predictions.
In addition, STGRNS has fewer hyperparameters compared to other GRN reconstruction methods based on deep learning models, which is one of the main reasons for its excellent generalization.
More information: Jing Xu et al, STGRNS: an interpretable transformer-based method for inferring gene regulatory networks from single-cell transcriptomic data, Bioinformatics (2023). DOI: 10.1093/bioinformatics/btad165
Journal information: Bioinformatics
Provided by Chinese Academy of Sciences