Massive single-cell atlas across human tissues highlights cell types where disease genes are active

Genetic studies have revealed many genes linked to both common and rare disease, but to understand how those genes bring about disease and use those insights to help develop therapies, scientists need to know where they are active in the body. Research on single cells can help achieve this goal, by surveying gene activity in specific cell types. Scientists need to profile all cell types and compare them across organs in the body to learn about the full range of human diseases, but this is difficult to do with existing methods.
Now researchers at the Broad Institute of MIT and Harvard have developed a robust experimental pipeline that can profile many more cell types from more tissues than can be studied with other techniques, as well as machine learning methods to put this data together and query the resulting map, or atlas. The team used it to pinpoint specific cell types from various tissues involved in multiple diseases. Their approach will enable other large-scale studies of diverse cell types and comparisons across tissues, including cells from frozen tissue that can be collected from many patients. This work opens up a wealth of samples stored in research collections around the globe for this kind of single-cell analysis, and also brings scientists a huge step closer towards their goal of a human cell atlas that catalogs every cell type in the human body, in a large number of individuals from diverse backgrounds.
Previous single-cell studies have mostly focused on one tissue type at a time, to create tissue-specific maps. Using their new pipeline, the team built a massive atlas of hundreds of thousands of cells across multiple tissues in the body. This allowed them to uncover unexpected new functions and gene expression programs for several cell types, such as muscle cell programs being expressed in lung connective tissue cells. The findings also revealed genetic similarities among cells in different tissues, and linked certain cell types to specific diseases for the first time.
The atlas is the first cross-tissue atlas to be based on measurements of gene activity within individual cell nuclei, which allowed the team to capture a greater variety of cell types than existing methods that measure gene expression from the whole cell.
The researchers say their atlas will spur many new studies on health and disease, and have openly shared it with the scientific community through the GTEx portal and the Broad's Single-Cell Portal.
This study is part of the international Human Cell Atlas (HCA) consortium, which is aiming to map every cell type in the human body as a basis for both understanding human health and for diagnosing, monitoring, and treating disease. An open, global, scientist-led consortium, HCA is a collaborative effort of researchers, institutes, and funders worldwide, with more than 2,300 members from 83 countries across the globe.
The paper is one of four major collaborative studies for the Human Cell Atlas published in Science this week, which have created comprehensive and openly available cross-tissue cell atlases. The complementary studies shed light on health and disease, and will contribute towards a single Human Cell Atlas.
"These studies represent a key moment for single-cell research and the Human Cell Atlas," said Aviv Regev, co-senior author of the study who was a core institute member at the Broad when the study began and is currently head of Genentech Research and Early Development. "In our study, we've shown that this approach can generate crucial insights about the role of cells and tissues in many diseases, which will spark new scientific and biomedical inquiries aimed at a shared goal of revolutionizing medicine."
The right teams at the right time
Over the last decade, Regev and others in the Klarman Cell Observatory at the Broad have been leaders in developing and implementing techniques that analyze the gene activity, or RNA expression, within individual cells, but those methods don't work well on large cells from fat or muscle tissues or on delicate cells like neurons. So scientists in the Regev lab began developing new approaches that could be applied to a wider variety of cell types by isolating the cell's nucleus for RNA measurement, rather than the entire cell. In addition, these approaches can conveniently be applied to frozen, rather than fresh tissue, which will enable researchers to collect the large numbers of samples needed to capture a diversity of human populations around the globe.
In parallel, another group of Broad scientists realized they would benefit from that same method. Broad researchers with the Genotype-Tissue Expression (GTEx) project, funded by the National Institutes of Health, had been documenting how small changes in DNA sequence, including disease-associated variants, can impact gene expression across dozens of tissues in the human body. Since 2010, they've analyzed dozens of tissue types from hundreds of donors using methods that process tissue into a bulk mixture, but they wanted to see how genetic variation altered individual cells.
"We needed a more precise look at cells within tissues, because the cell is where biology happens, both in health and disease," said institute scientist Kristin Ardlie, co-senior author on the new study and director of the GTEx Laboratory Data Analysis and Coordination Center at the Broad.
Existing single-cell RNA sequencing methods can be used to analyze fresh tissues, but the samples in GTEx's tissue bank were all frozen. Ardlie and her team suspected that the single-nucleus methods being developed in Regev's lab could give them a powerful way to analyze their banked frozen samples—and more cell types within them—while providing their colleagues with a comprehensive collection of human tissues they could use to benchmark the single-nucleus approach.
"The two groups needed each other, at the right time, to build a novel way of scaling up these studies," said study co-first author Gökcen Eraslan, a postdoctoral fellow at Genentech who was a member of the Klarman Cell Observatory when the study began.
Charting a new kind of cell atlas
In the new study, the GTEx team, the Regev lab, and their colleagues collaborated to develop a new large-scale single-nucleus sequencing pipeline. In an effort led by Orit Rozenblatt-Rosen, executive director of cell and tissue genomics at Genentech who was scientific director of the Klarman Cell Observatory during the study, the team first optimized four different single-nucleus protocols and then used them to analyze 200,000 cells in frozen samples of 8 tissue types that were initially collected by the GTEx project. They employed a deep-learning-based model to compare cell profiles across tissues, donors, and methods, and showed that their single-nucleus profiling pipeline performed as well as gold standard methods for measuring RNA in single cells, while capturing cell types that single-cell methods could not capture.
The researchers generated a cross-tissue molecular reference map that reveals critical data on the cell types residing in various tissues. "With these new technologies, we are able to chart cells across healthy tissues in the human body," said Rozenblatt-Rosen. "Doing so gives us a comprehensive foundation for understanding what goes awry in disease."
The scientists also demonstrated that the approach can generate new biological insights, which may spark new studies linking the findings to health and disease. For example, in all tissues, the team observed two populations of a type of immune cell called macrophages: one population that performs an immune role and another that supports the tissue's function, with different proportions of each found in various tissues. The finding helps explain how tissues achieve self-regulated equilibrium, or homeostasis, and how a type of white blood cell called monocytes mature into macrophages with different functions. In the lung, they also observed connective tissue cells called fibroblasts that express gene programs typically associated with muscle cell function, suggesting a yet unappreciated role for these cells in the lung tissue.
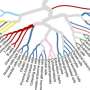
To explore the atlas's ability to support studies of disease, the team next turned to a catalog of Mendelian diseases, which are caused by changes to a single gene. The researchers cross-referenced the known 6,000 genes underlying these disorders with gene-level data from their atlas and identified new cell types that could be involved in disease, such as non-myocyte cell types that may play a role in muscular dystrophy. They also demonstrated the value of the atlas in proposing known and new cell types that may affect a range of common diseases and traits, like heart disease or inflammatory bowel disease, by comparing genes enriched in specific cell types to genes suggested by whole-genome association studies.
"Such cross-tissue cell atlases can help researchers understand the causes of comorbidities and how genetic variants can predispose to multiple diseases or conditions in the same person," said Ayellet Segrè, co-senior author of the study who is a Broad associate member and assistant professor at Mass. Eye and Ear and Harvard Medical School.
The researchers believe their approach now sets the stage for studies of greater scale, in hundreds of individuals or more from diverse ancestral backgrounds, to further explore the genes and cells underlying both rare and common diseases.
"Profiling multiple tissues is the only way to see this level of detail," said Eraslan. "We've always wanted to be able to profile the entire human body. In the past it's not been possible, but the technology and algorithms are mature enough to do this now. We've been waiting for this moment to come and now it's here."
The work was also led by Eugene Drokhlyansky, senior principal scientist at Bristol Myers Squibb who was a postdoctoral researcher at the Broad during the study, and François Aguet, a principal investigator at Illumina Artificial Intelligence Lab and former group leader in the Broad's Cancer Program.
More information: Gökcen Eraslan et al, Single-nucleus cross-tissue molecular reference maps towards understanding disease gene function, Science (2022). DOI: 10.1126/science.abl4290. www.science.org/doi/10.1126/science.abl4290
Journal information: Science
Provided by Broad Institute of MIT and Harvard