A new methodology for predicting corrosion rates
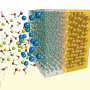
 form complex dynamic structures around dissolved metal ions (shown in green). The ease with which these structures can form or break apart often controls the rate at which metals corrode. Credit: Stephen Weitzner/LLNL")
Most metal alloys are prone to corrosion, which costs hundreds of billions of dollars of damage annually in the U.S. alone. Accurately predicting corrosion rates is a long-standing goal of corrosion science, but these rates depend strongly on the specific operating environment. At the atomic scale, these environmental factors are associated with how quickly and easily metal ions dissolve and transport across solid-liquid interfaces.
Lawrence Livermore National Laboratory (LLNL) scientists have used molecular dynamics simulations to unveil and describe the dynamical behavior of dissolved metal ions and water—a key component of this corrosion puzzle. They introduced a new methodology to describe the strength and nature of chemical bonding between rapidly moving ions in solution. These ions are surrounded by closely held water molecules in the so-called hydration shell, which fluctuates dynamically in ways that can be analyzed computationally. The authors presented a recipe for how these fluctuations could be quantified to ultimately develop computational "descriptors" for the propensity of metals to dissolve in harsh environments. The research appears in in The Journal of Physical Chemistry Letters.
Beyond corrosion, fluctuations in the hydration shell dictate critical processes in interfacial phenomena relevant to a broad array of applications, including water desalination and crystallization, as well as electrochemical energy storage and conversion.
The team came up with three metrics to represent the dynamical softness of ion hydration shells in terms of their rigidity, deformability and fluidity.
"Directly simulating ion transport across heterogenous interfaces remains technically challenging," said LLNL materials scientist Stephen Weitzner, lead author of the paper. "In many instances, we need to rapidly and accurately compare the effects of different environmental conditions on ion transport processes, but current simulation strategies can be too resource-intensive for high-throughput studies. So, we developed a set of dynamic metrics that provide a fast and intuitive way to compare the relevant chemical features.
"Our analysis showed that the new set of dynamic metrics not only correctly encoded key physical behavior, but also could explain trends in ion behavior that were challenging to classify using conventional static descriptors."
Weitzner added that beyond aiding the description and discussion of ion transport kinetics, the metrics provide useful targets for the development of machine learning-based force fields that could dramatically accelerate future simulations of metal ion dissolution and transport rates without loss of accuracy.
Other Livermore researchers include Tuan Anh Pham, Christine Orme, S. Roger Qiu and Brandon Wood.
More information: Stephen E. Weitzner et al, Beyond Thermodynamics: Assessing the Dynamical Softness of Hydrated Ions from First Principles, The Journal of Physical Chemistry Letters (2021). DOI: 10.1021/acs.jpclett.1c03314
Journal information: Journal of Physical Chemistry Letters
Provided by Lawrence Livermore National Laboratory