How SNPs can be used to detect disease pathways

Researchers at the University of Illinois Urbana-Champaign have developed a new computational tool that can identify pathways related to diseases, including breast and prostate cancer, using single-nucleotide polymorphisms. SNPs, which refer to mutations in a person's DNA, are the most common type of genetic variation among people. The researchers hope that the tool can help them discover new pathways that have been previously overlooked.
"This work was a part of the Mayo Grand Challenge, which aimed at improving our understanding of Hypoplastic Left Heart Syndrome," said Saurabh Sinha (BSD/CABBI/GNDP/GSP), a professor of computer science and the IGB's Director of Computational Genomics. "It is a rare, congenital heart disease that affects children and there is no cure. Our collaborators at the Mayo Clinic had sequenced the DNA of the children and their parents, and our colleagues at UIUC had identified mutations that were present in the children but not the parents. After that, we developed a tool to analyze the data to understand the disease pathways better."
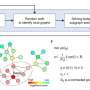
The tool, called VarSAn (Variant Set Annotator, pronounced 'version'), uses SNPs that have been identified by sequencing studies as being disease-related, to predict which pathways may be perturbed by these SNPs. Previously, scientists have looked at SNPs that show the strongest statistical signals with respect to a disease. They then carried out experiments to check whether each individual SNP was important.
"We're trying to approach the problem from a computational point of view. Does the whole gamut of SNPs identified in a genetic study point us to specific pathways that may not be known in the literature?" Sinha said. "The underlying computation is similar to how Google uses an algorithm to identify the right web pages for searches. These types of algorithms are applicable in biology as well to understand genetic variation. Additionally, 90% of the disease-related mutations are in parts of the DNA that do not code for proteins and using this type of approach will be useful for future work."
The VarSAn tool was validated by two distinct approaches. "We first did a literature search to see if those identified pathways were relevant to the diseases we were looking at," said Xiaoman Xie, a graduate student in the Sinha lab. "However, this type of validation is subjective and so we also developed an objective approach."
The second approach is based on testing the consistency of VarSAn's findings. "If we have two studies that identify two sets of SNPs associated with the same disease, the algorithm should ideally identify the same set of pathways for both of them i.e., it should be consistent. However, if the tool is given two sets of SNPs associated with two different diseases, it should report pathways specific to each disease but not the same set of pathways," Xie said.
The team is now trying to make VarSAn an online tool where researchers can paste the list of SNPs and the tool reports the pathways directly. "Currently, if a user wants to use this tool, they have to download the repository and run the code themselves, which can be inconvenient. We're working on making it easier," Xie said.
The work was funded by the Mayo Clinic Center for Individualized Medicine and the Todd and Karen Waneck Program for Hypolastic Left Heart Syndrome and the National Institutes of Health.
The study "VarSAn: associating pathways with a set of genomic variants using network analysis" was published in Nucleic Acids Research.
More information: Xiaoman Xie et al, VarSAn: associating pathways with a set of genomic variants using network analysis, Nucleic Acids Research (2021). DOI: 10.1093/nar/gkab624
Journal information: Nucleic Acids Research
Provided by University of Illinois at Urbana-Champaign