Molecular iodine catalyzes processes for antiviral and pharmaceutical syntheses

Synthesizing pharmaceuticals for cancer, viral diseases, and other medical conditions is slow work. A particularly challenging chemical transformation is to start with what's known as an unactivated alkene—a common molecular building block—and end up with a vicinal diamine; i.e., installation of two nitrogen units into carbon—carbon double bonds. The result is a chemical unit that's present in medications for influenza and colorectal cancer.
Commonly, researchers must use rare, toxic metals and harsh reaction conditions to complete this transformation. Using a more sustainable catalyst for the reaction could solve such problems. Previous research has attempted to do so, yet with only limited success.
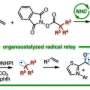
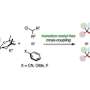
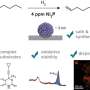
In a study recently published in the Journal of the American Chemical Society, researchers from Osaka University synthesized vicinal diamines from unactivated alkenes, using iodine as the catalyst. The synthetic protocol, appropriate for both anti- and syn-addition, is realistic, useful, and environmentally friendly.
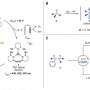
"We synthesized all diastereomers of vicinal diamines by anti-addition—adding two substituents to opposite sides of the double bond," says Satoshi Minakata, lead and senior author. "In the presence of a molecular iodine catalyst, unactivated alkenes reacted with commercially available nosylamide and sodium hypochlorite, to yield the intended products in a stereospecific manner."
The reactions were complete within 12 hours at only 40C for many types of cyclic and terminal alkenes, such as styrene derivatives. Diaminating an internal alkene with precise control of the three-dimensional shape of the final reaction products—important in many drug molecules—required only minor adjustments to the reaction temperature.
"Syn-addition—adding one or more substituents to the same sides of the double bond—required a different yet still mild reaction protocol," says Hayato Miwa, second author. "The alkene substrate scope for syn-addition was broad: we even fused heteroaromatic compounds across the bond."
A common limitation of previous vicinal diamine syntheses is the last step: removal of protecting groups—rather inert chemical units that mask the chemistry of one or more chemical units in the molecule. Protecting groups prevent otherwise reactive units in the molecule from interfering with the reaction at hand. The Osaka University researchers found that removing the protecting groups from the amines at the end of their syntheses was straightforward.
"The main byproducts of our protocol are sodium chloride and water," says Minakata. "We are doing our best to minimize the environmental impact of an important chemical reaction."
By using molecular iodine as the catalyst, instead of a toxic or rare metal, Minakata and coworkers are advancing the sustainability of pharmaceutical syntheses for future generations. Their approach will also help minimize possible chemical supply chain disruptions over the course of the ongoing pandemic.
More information: Satoshi Minakata et al. Diastereodivergent Intermolecular 1,2-Diamination of Unactivated Alkenes Enabled by Iodine Catalysis, Journal of the American Chemical Society (2021). DOI: 10.1021/jacs.1c00228
Journal information: Journal of the American Chemical Society
Provided by Osaka University