From molecule to medicine via machine learning

It typically takes many years of experiments to develop a new medicine. Although vaccines to protect against disease from the novel coronavirus are starting to reach clinics around the world, patients and doctors will still need treatments to manage COVID-19 symptoms for some time.
At Pacific Northwest National Laboratory (PNNL), computational biologists, structural biologists, and analytical chemists are using their expertise to safely accelerate the design step of the COVID-19 drug discovery process.
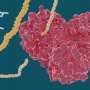
Rather than finding a new drug by trial and error, scientists are taking the three-dimensional structures of proteins from the novel coronavirus and using computer modeling and machine learning to identify a unique molecule that best fits inside a binding pocket on a protein's surface. Ideally, that molecule clogs the viral protein and prevents it from functioning.
"Drug research and development is a complex, costly, and time-consuming process, particularly considering the majority of molecules advanced from the design phase fail in clinical trials," said PNNL computational data scientist Neeraj Kumar. "Computer-based screening incorporates chemical information during the design process to increase a drug candidate's potential for success in clinical testing."
Developing an approach to speed drug discovery during this pandemic could also reveal new design steps that might be useful during the next outbreak.
Clogging coronavirus proteins
There are almost 30 different proteins in this novel coronavirus that are potential targets for COVID-19 drug discovery. Combine that with millions of molecules that are potential drug candidates, and the possibilities for matching molecules to specific proteins are mind-boggling.
To narrow the options towards molecules with potential to become medicines, Kumar and his team first use molecular docking to virtually screen libraries of known molecules and regulatory-approved drugs. Ones that fit in the binding pocket of a particular coronavirus protein make the short list for the next step of the process: testing the fit with actual proteins and molecules.
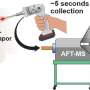
Experimental scientists then combine the molecules on this short list with purified coronavirus protein and "weigh them" with native mass spectrometry to see if the protein picked up the molecule. This technique measures interactions between the protein and the molecules and can confirm the predicted binding.
Quantifying how well the molecules bind to a protein is the next step. This provides critical information that helps scientists identify which ones might be the best candidates to carry forward in development.
That's where artificial intelligence helps. Molecular modeling and high-level quantum mechanical calculations generate a collection of properties of the protein-molecule complex. Machine learning algorithms identify patterns in those properties linked to binding. The result is a ranking of molecules based on predicted binding strength to a protein.
Kumar and his group are looking at molecules that rest in the binding pocket of some coronavirus proteins and prevent them from functioning, which is a common approach to drug development. In a less common approach called covalent inhibitor design, they are not only looking for molecules that fit into binding pockets, but also ones that form an irreversible chemical bond with an atom in the binding site. Drugs designed with this approach can have longer-lasting effects since they are physically connected to a protein.
The team's work is part of the U.S. Department of Energy's National Virtual Biotechnology Laboratory, a consortium of DOE national laboratories focused on response to COVID-19, with funding provided by the Coronavirus CARES Act.
Design, build, test, repeat
Once Kumar and his colleagues identify a promising candidate for further development, they send the molecular structure to National Virtual Biotechnology Laboratory colleagues who synthesize it for further testing.
Back at PNNL, analytical chemist Mowei Zhou performs some of those tests using mass spectrometry capabilities at the Environmental Molecular Sciences Laboratory, a DOE Office of Science user facility at PNNL. He combines the molecule with a purified coronavirus protein and looks for the "weight gain" of the protein due to binding of the molecule using native mass spectrometry.
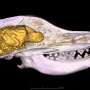
Structural biologist Garry Buchko then attempts to solve a structure for a protein-molecule complex with atomic level resolution. This provides structural details Kumar can use to refine the next round of computer modeling and further optimize the structure of the drug candidate.
Shape, fit, and binding strength are important steps in designing a new drug, although those features do not always correlate to how a drug functions in the body. Kumar and his colleagues also plan to build a machine learning model to predict properties related to how a drug travels through the body and gets metabolized along the way. That information can provide clues to potential toxicity or side effects in clinical trials.
"We hope the combination of structural design and activity predictions aided by machine learning can one day help speed the process of drug discovery in general," Kumar said.
Provided by Pacific Northwest National Laboratory