Mining molecular data with cryo-EM unveils hidden biological secrets

The field of structural biology has made enormous strides, peering into the activities of nature at the tiniest scale. Such investigations are critical for charting the behavior of important macromolecules and understanding their essential role in living organisms.
Researchers at the Biodesign Center for Applied Structural Discovery and ASU's School of Molecular Sciences have taken a new approach to studying molecules of life, examining not only their static structures at high resolution but the all-important dynamic movements of such molecules as they carry out biological functions.
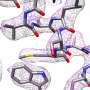
The new method involves an aggressive reprocessing of data obtained through a groundbreaking technique known as cryogenic electron microscopy or cryo-EM. Here, molecules targeted for study are flash-frozen in a thin membrane of ice before being subjected to electron microscopy. Tens or even hundreds of thousands of still images are collected, then reassembled by means of computer.
The technique offers a powerful alternative to X-ray crystallography for probing the molecular world in keen detail. Indeed, cryo-EM excels in the areas of study that are most challenging for X-ray crystallography, the imaging of large protein complexes resistant to conventional crystallization methods.
Although early iterations of cryo-EM struggled to compete with the extreme image resolution characteristic of X-ray crystallography, rapid advances in the field now enable cryo-EM to produce stunning macromolecular images at near-atomic-resolution.
In the new study, Abhishek Singharoy and his colleagues demonstrate that cryo-EM can be pushed to even greater extremes of clarity, by extracting precious information previously buried in the reams of cryo-EM data.
"Now, we can actually see minimum free-energy pathways image-by-image during a simulation," Singharoy says. "It was impossible to see energetically feasible molecular movies before. Now cryo-EM, machine learning and molecular dynamics simulations have got us there."
Abhishek is joined by joint first authors Ali Dashti and Ghoncheh Mashayekhi of the Department of Physics, University of Wisconsin Milwaukee and ASU researcher Mrinal Shekhar. The new study is the result of a collaboration between five groups: Abbas Ourmazd's, and Peter Schwander's at the University of Wisconsin in Milwaukee, Joachim Frank's at Columbia Medical Center, Amedee des Georges at CUNY, and Singharoy at ASU.
The findings are reported in the current issue of the journal Nature Communications.
Applying the new strategy pioneered by co-authors Abbas Ourmazd and 2017 Chemistry Nobel Laureate Joachim Frank, which involves mathematical techniques of geometric machine learning combined with classical molecular dynamics simulations, helped researchers capture the fleeting movements of ryanodine receptor type 1, an important calcium channel able to bind other molecules. Subtle conformational changes of the receptor play a crucial role in the contraction of skeletal muscle and muscles of the heart, once the receptor has been triggered by a specific binding molecule.
Using single-particle cryo-EM, the group was able to assemble impressive molecular movies of ryanodine receptor type 1's continuous conformational changes, built from some 800,000 cryo-EM snapshots of molecules trapped in ice, like insects entombed in amber.
Combining snapshots that were intermediary between the fully closed and open conformations helped capture this receptor's structural shape-shifting before and after binding by activating molecules.
The new technique will be a boon in practical areas, particularly, drug discovery, while helping to resolve foundational issues in molecular biology.
More information: Ali Dashti et al, Retrieving functional pathways of biomolecules from single-particle snapshots, Nature Communications (2020). DOI: 10.1038/s41467-020-18403-x
Journal information: Nature Communications
Provided by Arizona State University